Tetralogy of Fallot
Tetralogy of Fallot is a congenital heart anomaly that can be associated with underlying chromosomal conditions.
Overview
Tetralogy of Fallot (ToF) is characterised by a combination of four specific heart anomalies. It can be detected antenatally or in the neonatal period by echocardiogram (cardiac ultrasound).
Clinical features
The clinical features of ToF are listed below. For a comparison with a normal heart, see figure 1.
- Overriding aorta: the aorta is positioned directly over a ventricular septal anomaly, instead of over the left ventricle.
- Pulmonary stenosis: a narrowing of the valve located between the right ventricle and the pulmonary arteries.
- Ventricular septal anomaly: a hole in the septum that separates the two ventricles of the heart.
- Right ventricular hypertrophy: an atypical enlargement in muscle mass of the right ventricle.
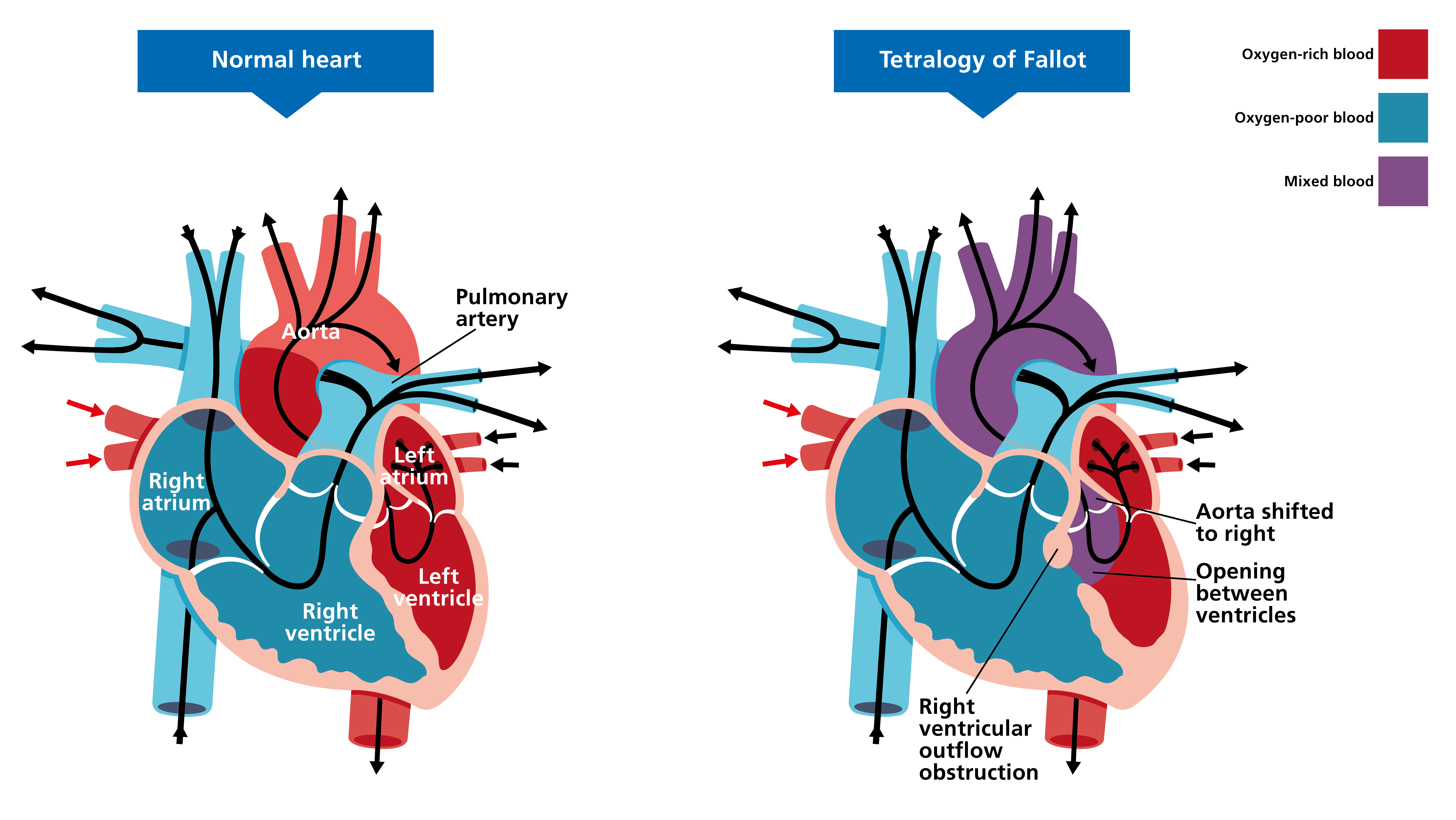
Figure 1: Clinical features of tetralogy of Fallot
Presenting features
Patients with ToF will present with some or all of the following symptoms:
- cyanosis;
- shortness of breath and rapid breathing, especially during feeding or exercise;
- floppy and/or pale appearance;
- poor weight gain;
- irritability;
- prolonged crying;
- heart murmur; and
- an atypical, rounded shape of the nail bed in the fingers and toes (clubbing).
Please note that symptoms vary, depending on the amount of blood flow.
Genomics
No genetic cause has been identified for the majority of ToF cases (though some patients may have additional anomalies and/or health issues that are part of a genetic syndrome). It should be noted, however, that children with ToF will have an increased chance of having chromosomal conditions – see the following examples.
- 22q11.2 deletion syndrome: Also known as DiGeorge syndrome, this is caused by a microdeletion at chromosome position 22q11.2, which can be found via microarray, fluorescence in situ hybridisation (FISH) or targeted gene testing. Around 75% of patients with 22q11.2 deletion syndrome will have symptomatic congenital heart disease, of which ToF is one of the most common.
- Down syndrome: This condition is caused by an extra copy of chromosome 21 (trisomy 21), and can be found via common aneuploidy testing (QF-PCR). ToF occurs in around 6% of patients with Down syndrome and is the most common cyanotic heart anomaly to present in this patient group. Conversely, around 8% of patients with ToF will have Down syndrome.
- Alagille syndrome: This condition is caused by variants in either the JAG1 gene or the NOTCH2 gene, and is found via microarray, FISH or targeted gene testing. Around 90% of children with Alagille syndrome will have a heart anomaly, around 12% of which will be ToF.
Diagnosis
ToF is diagnosed via cardiac imaging (typically an echocardiogram/ECHO) which may be carried out for a variety of reasons. In the prenatal setting, the cardiac structures are assessed at the routine 20 week scan; however, in a significant minority of cases, the anomaly is not picked up during this scan and may then present postnatally. Postnatally, signs such as a murmur, poor feeding, irritability or cyanosis may prompt further investigations. Additional signs and symptoms such as dysmorphism, additional congenital anomalies or developmental delay may point towards a specific underlying genetic diagnosis.
For more information about genetic testing in the context of pregnancy, please see Fetus with anomalies discovered during the 20-week screening scan. For information about genetic testing after birth, please see Infant or child with congenital heart disease.
Inheritance and genetic counselling
Isolated ToF has a multifactorial mode of inheritance in most cases, and recurrence risk rates of 2.5%–3% have been attributed to first-degree relatives of an affected child. In a subgroup of patients with a strong family history, the transmission of a monogenic trait has been suspected.
If you are discussing genomics concepts with your patients, you may find it helpful to use the visual communication aids for genomics conversations.
Management
Treatment comes in the form of major cardiac surgery, usually at around four to six months of age. Without the surgery, most children would sadly not live to adulthood. Dependent on the individual clinical picture, a shunt may be needed prior to the surgery. The majority of children will survive the surgery and have a good quality of life. In rare cases, further surgeries are required, and ongoing care from a cardiology team is recommended.
Resources
For clinicians
- Leeds Congenital Heart Unit: Tetralogy of Fallot (video, 3 minutes 12 seconds)
- NHS England: National Genomic Test Directory
- The Fetal Medicine Foundation: 2nd and 3rd trimester: Tetralogy of Fallot
For patients
- British Heart Foundation: Tetralogy of Fallot
- International Society of Ultrasound in Obstetrics and Gynaecology (ISUOG): Tetralogy of Fallot