Myotonic dystrophy
Myotonic dystrophy is a highly variable genetic condition that causes progressive muscle loss and weakness.
Overview
Myotonic dystrophy (DM) is an autosomal dominant, multisystem disease with a common pattern of clinical signs and symptoms including myotonia, muscular dystrophy, cardiac conduction anomalies, cataracts and endocrine conditions. There are two types of DM: type 1 (DM1) and type 2 (DM2). DM1 tends to be more severe than DM2 and is more common in the UK.
Clinical features
The age at which symptoms start is highly variable: they can present at any time from birth to old age. In general, the later the condition starts, the milder it will be.
Key features include:
- progressive muscle weakness and wasting;
- myotonia;
- dizziness and syncope due to heart conduction dysfunction (arrhythmias and conduction blocks);
- swallowing difficulties;
- cataracts;
- learning difficulties in some affected children;
- hypersomnia;
- frontal balding;
- gastrointestinal conditions;
- insulin resistance and diabetes mellitus; and
- thyroid dysfunction.
The two types of DM are characterised by the affected gene, age of onset and clinical presentation.
Myotonic dystrophy type 1 (DM1)
- Affected gene: DMPK (chromosome 19), CTG repeat expansion.
- Age of onset: Usually during adulthood, though there is a childhood form and a congenital form.
- Presentation:
- In children and adults:
- facial weakness, ptosis and distal muscle weakness (foot drop or gait disturbance);
- grip myotonia is common; however, myotonia may affect any other muscle, including bulbar, tongue or facial muscles, causing problems with talking, chewing, and swallowing;
- arrhythmias and conduction blocks; and
- serum creatine kinase (CK) may be mildly elevated in those with weakness, though may be normal in asymptomatic individuals.
- In congenital forms:
- hypotonia and paucity of movements;
- common respiratory issues, some needing mechanical ventilation;
- weak cry and inability to suck;
- facial diplegia with tent-shaped upper lip and high arched palate;
- very high serum CK;
- intellectual disability (50%–60% of cases); and
- low visual acuity and hyperopia.
- In children and adults:
Myotonic dystrophy type 2 (DM2)
- Affected gene: CNBP (chromosome 3), CCTG repeat expansion.
- Age of onset: Adult onset only (typically 30s to 40s).
- Presentation:
- exercise-induced fatigue and myalgia and/or mild grip myotonia and myotonia of the proximal legs;
- difficulty rising from a chair or a squat, and difficulty climbing stairs; and
- mild to moderate serum CK elevation, although high serum CK elevation can be found.
Genomics
Both types of DM are nucleotide repeat expansion disorders, caused by the pathological expansion of intragenic tri-/tetra-nucleotide repeats. DM1 is caused by an atypical number of CTG repeats in the DMPK gene, and DM2 by an atypical number of CCTG repeats in the CNBP gene.
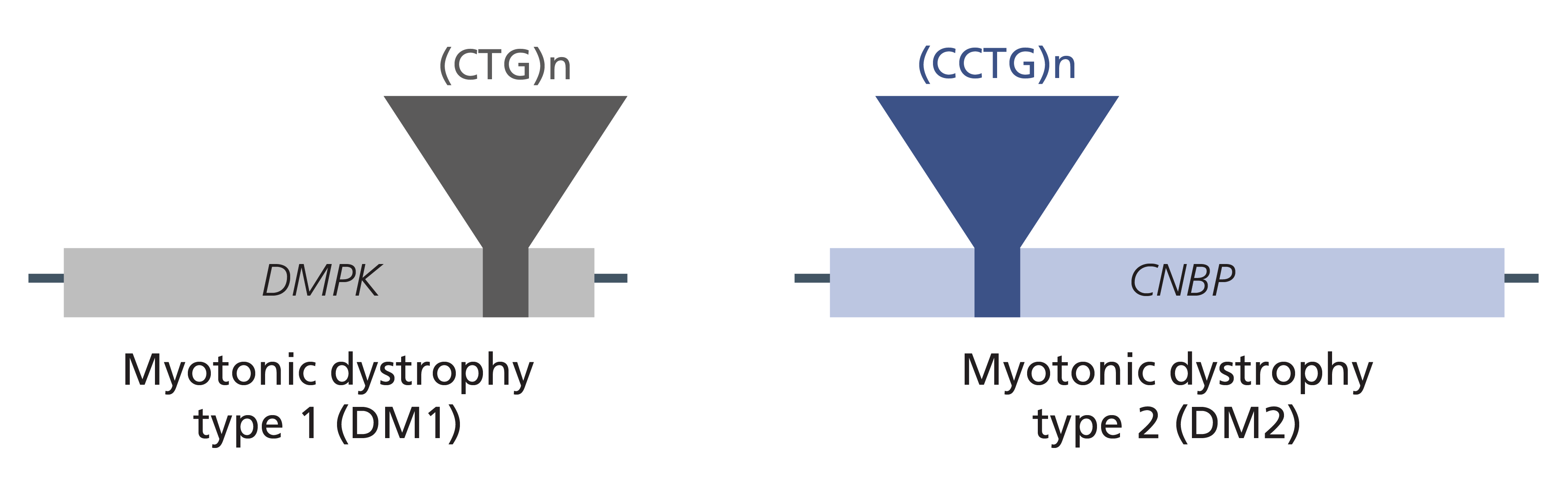
Figure 1: Genes and variants associated with myotonic dystrophy.
Unlike other disease-causing pathogenic variants (such as single nucleotide changes), which are usually passed unchanged from parent to child, nucleotide repeat expansions are considered ‘dynamic’ due to their tendency to expand further during DNA replication. This explains the phenomenon observed in DM1 (and several other nucleotide repeat expansion disorders, but not DM2) called ‘anticipation’, which describes the tendency of the disease phenotype to become more severe and/or earlier in onset in successive generations.
The longer a repeat region becomes, the more inherently unstable it is. When an individual has a normal number of repeats in the gene it is relatively stable and does not have a tendency to expand when passed to the next generation (5–34 repeats in DM1; 11–26 repeats in DM2). However, there is a threshold for instability, and when the number of repeats falls within the ‘premutation’ range (35–49 repeats in DM1; not found in DM2), there is a chance of expansion upon transmission from parent to child. The pathological range for repeats is 50–5,000 in DM1 (classic: 100–1,000; congenital: greater than 1,000) and 75–11,000 in DM2.
Diagnosis
Diagnosis of DM is based on personal and family history as well as examination findings. Diagnosis may be supported by blood tests (including serum creatine kinase (CK)) and electromyography (EMG) studies, and confirmed by genomic testing. For information about genomic testing for DM, see Presentation: Hypotonic infant and Presentation: Neonate with suspected myotonic dystrophy type 1.
Inheritance and genomic counselling
Both types of DM are autosomal dominant conditions.
Genomic counselling in DM1
Genomic counselling in DM1 is complex, due to the possibility of expansion of the nucleotide repeat as it is passed from parent to child.
- An individual with DM1 has almost invariably inherited the expanded repeat from a parent with a CTG repeat number of over 34 (as new expansions of a gene with repeat size in the normal range into the atypical range are rare). Therefore, even asymptomatic parents of a proband should be offered genomic testing, where appropriate, to define how likely it is that family members or future offspring will be or have been affected.
- If one parent has an expanded DMPK allele, the chance of future offspring inheriting the condition is 50%.
- Note that the allele may expand further in length on transmission (see above), so parents should be counselled that there is a tendency for offspring to be affected with earlier onset and more severe disease than the parent.
- Additionally, there is a gender effect on the likelihood of transmission. The triplet repeat is more prone to expansion when passed on by the mother than by the father, with the most severe congenital DM most commonly (but not invariably) resulting from maternal transmission.
- Prenatal and/or preimplantation genetic diagnosis may be considered for families who could be affected.
Genomic counselling in DM2
Genomic counselling in DM2 is more straightforward because, unlike in DM1, anticipation is not observed.
- An individual with DM2 has inherited the repeat from a parent with a CNBP repeat expansion. (Probands with de novo pathogenic variants have not been reported and ‘premutation’ alleles are not observed in DM2).
- If it is not possible to establish which parent is affected using family history or examination, molecular genomic testing is appropriate.
- Where a parent of the proband is affected or found to have an expanded CNBP repeat expansion, the chance of future offspring inheriting the condition is 50%.
- Anticipation is not observed in DM2.
- Predictive (also known as pre-symptomatic) genomic testing may be relevant for wider (adult) family members, and these individuals may be referred to their local genetics service. In general, testing is not offered to unaffected children younger than 18 years of age, because symptom onset typically occurs in adulthood and there is no preventative or early intervention management that has been shown to improve outcomes.
Management
Management of children with DM is complex and should be delivered via a multidisciplinary team, including neurology, cardiology, ophthalmology, endocrinology and community paediatrics. Detailed suggested approaches have been published by several authors (see our resources list below).
Women with DM1 are at greater risk of pregnancy complications and should be monitored accordingly.
Resources
For clinicians
- GeneReviews: Myotonic Dystrophy Type 1
- GeneReviews: Myotonic Dystrophy Type 2
- Genomics England: NHS Genomic Medicine Service Signed Off Panels Resource
- NHS England: National Genomic Test Directory
- US National Library of Medicine: ClinicalTrials.gov database
References:
- Ashizawa T, Gagnon C, Groh WJ and others. ‘Consensus-based care recommendations for adults with myotonic dystrophy type 1’. Neurology Clinical Practice 2018: volume 8, issue 6, pages 507–520. DOI: 10.1212/CPJ.0000000000000531
- Turner C and Hilton-Jones D. ‘The myotonic dystrophies: diagnosis and management’. Journal of Neurology, Neurosurgery & Psychiatry 2010: volume 81, issue 4, pages 358–367. DOI: 10.1136/jnnp.2008.158261
For patients
- Muscular Dystrophy UK: Myotonic dystrophy