Fumarate hydratase tumour predisposition syndrome (including hereditary leiomyomatosis and renal cell cancer)
Fumarate hydratase tumour predisposition syndrome defines a group of overlapping conditions, including hereditary leiomyomatosis, renal cell cancer and fumarate hydratase-deficient renal cell carcinoma. It is caused by a constitutional (germline) pathogenic variant in one FH allele.
Gene function
The FH gene encodes the fumarate hydratase/fumarase (FH) protein, which catalyses the conversion of fumarate to malate in the tricarboxylic acid (Krebs) cycle (see figure 1).
Gene locus and structure
The FH gene is located at chromosome 1q43, and comprises 10 exons and 510 amino acids.
Prevalence
The population frequency of fumarate hydratase (FH) tumour predisposition syndrome is uncertain. While older estimates suggest a prevalence of 1 in 200,000, a recent analysis of non-population-based cohorts found a frequency of pathogenic variants in FH of 1 in 1,000 individuals. This suggests that there may be a higher number of cases displaying milder or no symptoms, reflecting variable expressivity and reduced penetrance.
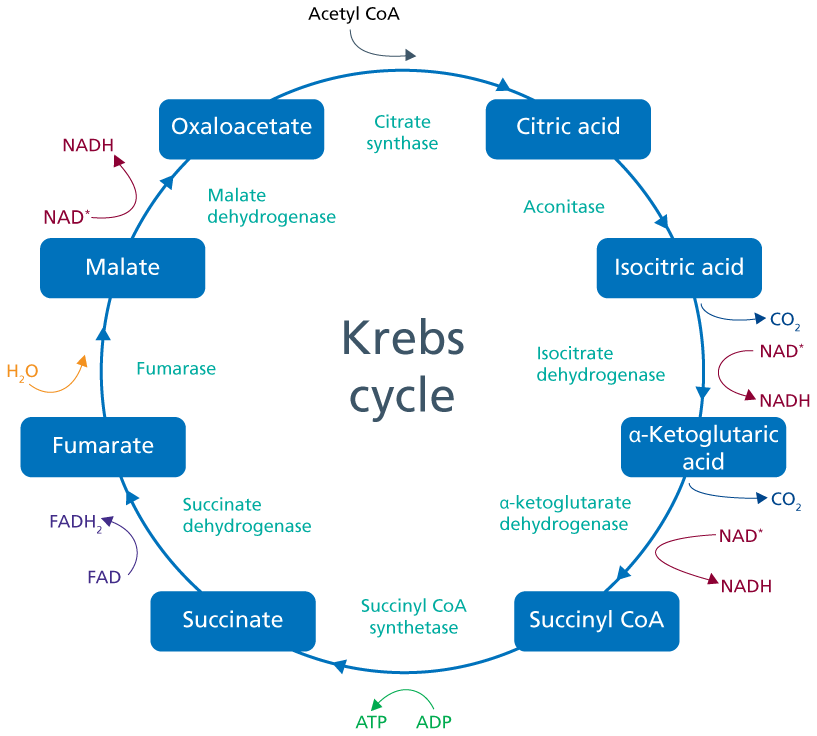
Figure 1: The Krebs cycle also known as the tricarboxylic acid cycle or citric acid cycle, takes place in the mitochondria and uses acetyl CoA as the starting substrate. A series of redox reactions facilitate production of cellular energy in the form of ATP. Image created in and exported from biorender.com under a paid subscription (Terri McVeigh).
Disease associations
FH tumour predisposition syndrome is typically characterised by the presence of at least one of a triad of phenotypic features, including cutaneous leiomyomas, uterine leiomyomas (fibroids) and renal cancers, which gave rise to the older term ‘hereditary leiomyomatosis and renal cell cancer syndrome’ (HLRCC). The prevalence of such phenotypic features in affected individuals is highly variable, and the term ‘FH tumour predisposition syndrome’, which encompasses presentations with isolated phenotypic features, is now preferred.
Clinical features
- Cutaneous leiomyomata: Firm skin-toned or slightly darker papules (single or clustered) frequently found on the trunk and extremities. These occur in up to 80% of individuals with HLRCC and are often noted to be painful or sensitive to touch. They frequently appear by the fourth or fifth decade of life and variably increase in size and number with age.
- Cutaneous leiomyomata are predominantly benign. Although malignant transformation to leiomyosarcoma has been reported, it is rare, and reports should be treated with caution: changes over time in nomenclature and diagnostic criteria mean that lesions previously called leiomyosarcoma may have, in fact, been atypical smooth-muscle neoplasms or leiomyomata.
- Uterine leiomyomata (uterine fibroids): These occur frequently in females with HLRCC (reports range from 40%–80%). They tend to develop at a young age (under 40 years) and are large and numerous, commonly causing irregular menses, pain and menorrhagia. A significant number of cases may require intervention with myomectomy or hysterectomy.
- When HLRCC was originally described, leiomyosarcoma was reported in a small number of women. It is unclear whether women with HLRCC have a higher risk of developing uterine leiomyosarcomas than women of a similar age in the general population. Most families are not highly predisposed to uterine cancer.
- Renal cell carcinoma (RCC): Affected individuals have a lifetime risk of developing RCC of up to 21%. It is typically characterised by solitary, unilateral tumours that are aggressive and metastasise early. There is a young age of onset compared with sporadic RCC (the mean age of diagnosis is 44 years, but the youngest reported case is 11 years).
- Affected individuals will have a particular predisposition to type two papillary RCC, and also to a spectrum of other pathologies that have been observed (including undefined papillary, clear cell, collecting duct and tubulocystic tumours).
- Phaeochromocytoma and paraganglioma: These are rarely reported in association with HLRCC (less than 1% cases). Specific lifetime risks are not known but are low, and dedicated surveillance is not indicated.
FH-deficient tumours (uterine fibroids and RCC) are histopathologically distinct. Hallmark cytological features include single or multinucleated nuclei with large inclusion-like eosinophilic and orangiophilic nucleoli surrounded by a clear halo. FH-deficient RCC is a WHO pathology definition. Cancers occurring within the context of FH tumour predisposition syndrome often show loss of FH staining and positive staining for S-(2-succino) cysteine on immunohistochemistry (though this is not routinely available).
Sporadic tumours with biallelic FH variants
Sporadic tumours have been found to have biallelic inactivating variants in FH that are not constitutional (germline). Although this is very rare, it needs to be considered in individuals presenting with an FH-deficient tumour in the absence of any other findings consistent with FH tumour predisposition syndrome. Careful phenotyping and consideration of constitutional mosaicism is required.
Recessive conditions associated with pathogenic variants in FH
FH deficiency is a rare autosomal recessive metabolic disease that occurs in individuals carrying pathogenic variants in both copies of the FH gene (usually inheriting one pathogenic variant from each parent). It presents as an early-onset severe encephalopathy with seizures, developmental delay and atypical brain development (including cerebral atrophy, enlarged ventricles, bilateral polymicrogyria and thinning or absence of the corpus callosum).
The risk of FH deficiency should be considered if a person with a heterozygous constitutional (germline) FH variant is in a consanguineous relationship. Outside of consanguineous relationships, the frequency of pathogenic variants in FH in the general population is low, such that NHS-funded carrier testing of partners and spouses is not typically recommended based on current guidelines.
Pathogenic variant spectrum
- FH tumour predisposition syndrome is an autosomal dominant condition, caused by heterozygous constitutional (germline) inactivating variants in the FH gene. A wide pathogenic variant spectrum has been described in affected patients, including missense, frameshift, nonsense and splice-site variants, as well as large deletions.
- There are no known discernible genotype-phenotype correlations for FH tumour predisposition syndrome, and there are no common founder variants within any specific population.
- Variants associated with reduced penetrance have been described, including the in-frame variant FH: c.1431_1433dup, p.(Lys477dup). This variant has been:
-
- reported, though rarely, in those with heterozygous variants with isolated features of FH tumour predisposition syndrome; however there is increasing evidence contradicting the association of this variant with disease risk in heterozygous individuals;
- seen at a relatively high frequency in the general population, including in two homozygous individuals; and
- identified in trans with a different pathogenic variant in individuals with FH deficiency. Further research is required to fully establish the impact of this variant.
Genomic testing
Single gene testing of FH can be requested using the National Genomic Test Directory test code R365 Fumarate hydratase-related tumour syndromes. Eligibility criteria include the presence of:
- type-two papillary, HLRCC-associated RCC (WHO pathology definition) or tubulo-papillary renal tumour at any age;
- two of the following:
- cutaneous leiomyomata;
- renal tumour (any histology);
- uterine leiomyomata with classic histological features; and/or
- FH deficiency (any age);
- cutaneous leiomyomata and one first-, second- or third-degree relative with renal tumour;
- cutaneous leiomyomata and two first-, second- or third-degree relatives with cutaneous leiomyomata, or uterine leiomyomata with classic histological features <40 years;
- uterine leiomyomata with classic histological features <40 years; or
- multiple cutaneous leiomyomata (histologically confirmed).
FH gene testing is also offered as part of a broader multi-gene panel for kidney cancer predisposition, which can be requested using test directory code R224 Inherited renal cancer. Eligibility criteria include:
- renal cancer ≤40 years;
- type two papillary renal cancer ≤50 years;
- bilateral or multifocal renal cancer at any age;
- renal cancer and a first- or second-degree relative with renal cancer, where both cases have been diagnosed <50 years; or
- renal cancer and features of inherited cancer syndrome.
Tumour-based testing of FH should be undertaken (using test directory code M18 Renal cell carcinoma – adult) where immunohistochemistry undertaken on RCC indicates FH deficiency but no constitutional (germline) variant in the FH gene has been identified.
At present, testing of FH does not include dosage analysis. A diagnosis of FH tumour predisposition syndrome may be made on a clinical basis in the absence of a molecular diagnosis in individuals with suspicious clinical features if no alternative somatic (tumour) aetiology is identified. Orthologous testing may be required to confirm the diagnosis in such cases, and they should be discussed at specialist multidisciplinary team meetings.
Genomic counselling
- Cancer risk associated with constitutional (germline) FH variants is inherited in an autosomal dominant pattern. In contrast, FH deficiency is inherited in an autosomal recessive pattern.
- First-degree relatives of an individual with an inherited pathogenic variant in FH are at 50% risk of inheriting the familial variant.
- Referral to clinical genetics should be arranged for newly identified individuals with pathogenic or likely pathogenic variants in FH, to discuss onward management, family planning implications and cascade testing of at-risk relatives.
- If you are discussing genomics concepts with your patients, you may find it helpful to use the visual communication aids for genomics conversations.
Risk-reducing strategies
- RCC occurring in the context of FH tumour predisposition syndrome is a particularly aggressive disease. As such, prompt surgical excision with wide-margin resection is advised.
- Screening strategies should be considered for individuals with a constitutional (germline) variant in FH, including:
-
- renal surveillance imaging, preferably by annual MRI with 1mm to 3mm slices, commencing from 10 years of age; and
- dermatological review as and when required.
- Note that screening for phaeochromocytoma and paraganglioma with plasma metadrenaline levels is not routinely undertaken, unless there is a family history of these tumour types.
- Cutaneous leiomyomata may not require treatment and dermatologists do not recommend routine surgical excision, as lesions can recur and it can lead to scarring. Pain control should be optimised. Medications such as vasodilators (such as nifedipine and doxazosin) and drugs for treatment of neuropathic pain (such as gabapentin and pregabalin) are more helpful than standard analgesics. Other options include cryotherapy or laser ablation. Occasionally, surgical excision of a single, painful cutaneous lesion may be appropriate, but this is best guided by specialist dermatologist input.
- Uterine leiomyomata may require treatment if it is symptomatic. Non-surgical options include analgesics and gonadotrophin-releasing medications; however, surgical management is often required, sometimes at a young age, and can involve myomectomy or full hysterectomy. This can have significant reproductive implications for women with FH-associated fibroids.
Family planning implications
The Human Fertilisation and Embryology Authority has approved the use of preimplantation genetic testing for monogenic disorders (PGT-M – previously known as pre-implantation genetic diagnosis) for couples in whom one or both parent(s) has a likely pathogenic or pathogenic variant in FH. It is best practice that discussions regarding PGT-M and other family planning options be undertaken by a specialist genetic counsellor or clinical geneticist.
Other options may include prenatal testing (invasive, or non-invasive if the intended father has the variant) with termination of affected embryos, adoption, gamete donation, or natural conception and pregnancy with testing of children later in life.
Resources
For clinicians
- GeneReviews: FH tumour predisposition syndrome
- NHS England: National Genomic Test Directory (note that somatic (tumour) tests are listed in the directory for cancer, while constitutional (germline) tests are listed in the directory for rare and inherited disease)
- WHO International Agency for Research on Cancer: Urinary and male genital tumours
References:
- Forde C, Lim DHK, Alwan Y and others. ‘Hereditary leiomyomatosis and renal cell cancer: Clinical, molecular, and screening features in a cohort of 185 affected individuals’. European Urology Oncology 2020: volume 3, issue 6, pages 764–772. DOI: 10.1016/j.euo.2019.11.002
- Muller M, Ferlicot S, Guillaud-Bataille M and others. ‘Reassessing the clinical spectrum associated with hereditary leiomyomatosis and renal cell carcinoma syndrome in French FH mutation carriers’. Clinical Genetics 2017: volume 92, issue 6, pages 606–615. DOI: 10.1111/cge.13014
- Schuch B, Li S, Risch H and others. ‘Estimation of the carrier frequency of fumarate hydratase alterations and implications for kidney cancer risk in hereditary leiomyomatosis and renal cancer’. Cancer 2020: volume 126, issue 16, pages 3,657–3,666. DOI: 10.1002/cncr.32914
- Thompson AJ, Alwan YM, Ramani VAC and others. ‘Cost-effectiveness model of renal cell carcinoma (RCC) surveillance in hereditary leiomyomatosis and renal cell carcinoma (HLRCC)’. Journal of Medical Genetics 2022: volume 60, issue 1, pages 41–47. DOI: 10.1136/jmedgenet-2021-108215
- Zhang L, Walsh MF, Jairam S and others. ‘Fumarate hydratase FH c.1431_1433dupAAA (p.Lys477dup) variant is not associated with cancer including renal cell carcinoma’. Human Mutation 2020: volume 41, issue 1, pages 103–109. DOI: 10.1002/humu.23900
For patients
- HLRCC Foundation
- Kidney Cancer UK
- National Cancer Institute: Hereditary kidney cancer syndromes
- UK Kidney Association: Inherited renal cancer syndromes