Chronic granulomatous disease
Chronic granulomatous disease is a group of genetic conditions characterised by recurrent infections and granuloma formation.
Overview
Chronic granulomatous disease (CGD) is a group of genetic conditions characterised by life-threatening bacterial and fungal infections and multi-system inflammatory granulomata. Pathogenesis results from defective oxidative bursts in phagocytes that normally help destroy pathogens. Pathogenic variants in genes that encode the various subunits of the NAPDH oxidase complex (responsible for the oxidative burst) cause different types of CGD.
Clinical features
CGD typically presents at two to three years of age, though the total range is from infancy to late adulthood.
Clinical features of CGD include:
- recurrent, persistent bacterial and fungal infections:
- commonly pneumonia, which may be complicated by empyema; and
- lymphadenitis;
- abscess;
- osteomyelitis; and
- cellulitis;
- skin (often perianal) and/or liver abscesses;
- inflammatory colitis;
- granulomata formation:
- typically genitourinary and gastrointestinal;
- papulopustular dermatitis; and
- growth restriction
Bacterial infections are typically with encapsulated organisms, such as Staphylococcus aureus, Serratia sp, Burkholderia sp and Nocardia sp. BCG vaccination may lead to localised disease and draining skin lesions.
Fungal infections (cutaneous or invasive) develop in around 30% of cases and may be insidious in presentation. Aspergillus sp is the most common cause of invasive fungal infections, typically in the lung.
X-linked CGD has the most significant disease course and lower survival rates. Female carriers of X-linked CGD may also be symptomatic, particularly with discoid photosensitive lupus erythematosus, skin ulcers and arthritis/arthralgia.
Infections may be difficult to treat and can cause significant complications.
Genomics
Different genes are associated with different causes of CGD (see table 1 below).
Table 1: Genes and associated causes of CGD
| Gene | Type | Protein | Inheritance | Proportion of cases |
| CYBB | X-linked | gp91phox | X-linked | >60% |
| NCF1 | Type 1 | p47phox | Autosomal recessive | 20% |
| NCF2 | Type 3 | p67phox | Autosomal recessive | <10% |
| NCF4 | Type 2 | p40phox | Autosomal recessive | <1% |
| CYBA | Type 4 | p22phox | Autosomal recessive | <10% |
| CYBC1 | Type 5 | Eros | Autosomal recessive | <1% |
The prevalence of CGD is about 1 in 200,000 people. X-linked CGD is the most commonly encountered form. Other causes are autosomal recessive.
Some genotype-phenotype correlations have been recognised. These include the below.
- Inflammatory bowel disease is especially severe and difficult to manage in cases caused by CYBC1 and NCF4 deficiency.
- Some hypomorphic variants that result in proteins with some residual function cause a milder disease course.
- In males, deletions, nonsense variants and missense variants in the region of CYBB responsible for superoxide function (FAD- and NADPH-binding domains) are highly deleterious and associated with poorer outcomes.
- Some males with X-linked CGD due to contiguous gene deletions have additional medical problems resulting from the loss of closely located genes at Xp21.2, including:
- XK: McLeod neuroacanthocytosis syndrome (a multisystem condition with degenerative neurological, neuromuscular, cardiovascular and haematological manifestations);
- RPGR: RPGR-related retinitis pigmentosa (night blindness and progressive visual impairment);
- DMD: Duchenne muscular dystrophy (progressive muscle weakness, high CK and cardiomyopathy); and
- OTC: ornithine transcarbamylase deficiency (faltering growth, lethargy, vomiting, seizures and developmental delay).
Figure 1: NADPH oxidase complex in phagocytes
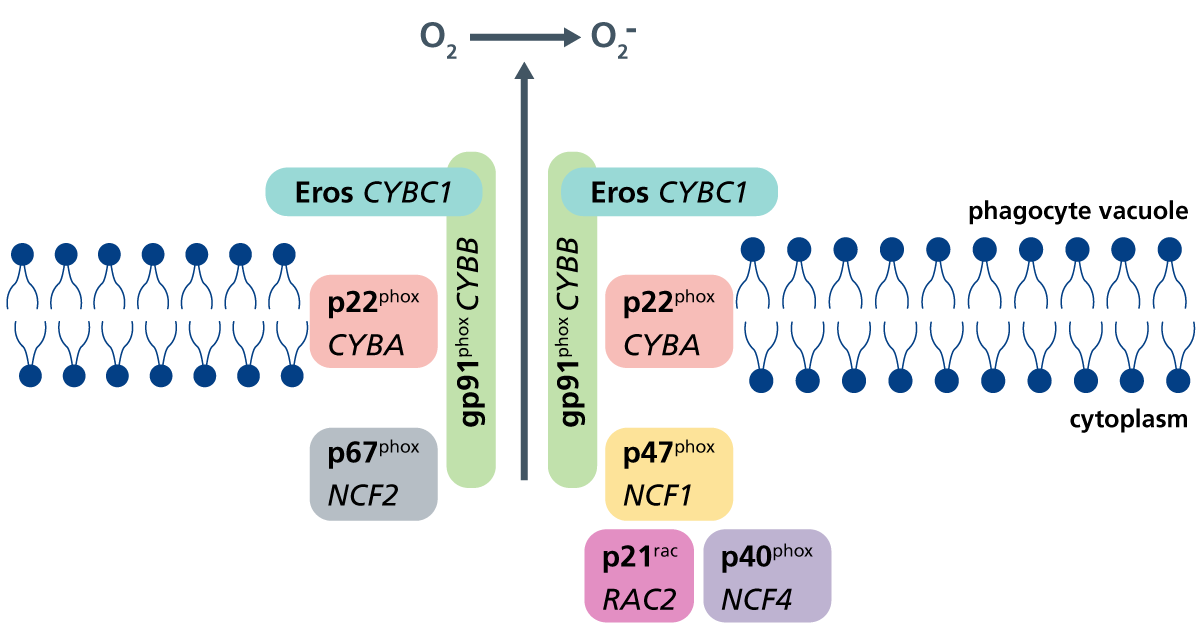
Defects in each of the subunits depicted cause susceptibility to recurrent bacterial and fungal infections. Genes encoding the individual proteins are shown in italics. (Click to enlarge in a new tab.)
Diagnosis
A clinical diagnosis of CGD may be made in patients presenting with suggestive clinical features, as listed above.
The following laboratory tests can help support the diagnosis:
- Nitroblue tetrazolium (NBT) and dihydrorhodamine (DHR) tests, which measure neutrophil superoxide production via the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex; and
- in males, gp91phox expression (protein affected in the X-linked form of CGD) may be useful.
Molecular diagnosis is made through the identification of pathogenic variant(s) in one of six genes that encode or permit assembly of the subunits of phagocyte NADPH oxidase.
For information about testing, see ‘Infant or child with severe, recurrent, persistent and/or unusual infections‘. If a specific diagnosis of CGD is suspected, this should be made clear on the request form, along with any supportive clinical and laboratory features, to aid with variant interpretation.
CGD may be identified before any symptoms appear, for example through the Generation Study. Types 2, 3, 4 and 5, as well as X-linked CGD, are included in the study. Confirmation of the diagnosis will require referral to clinical immunology services. Please refer to the local pathway for your region for this condition.
Once a genetic diagnosis has been identified, there are some additional considerations for certain genetic causes. These are outlined below.
- NBT and DHR testing may be equivocal in types 3 to 5 CGD (NCF4, CYBA and CYBC1 respectively). Where genetic analysis suggests this diagnosis, functional confirmation should be sought.
- NCF1 and NCF2 both have pseudogenes close by, which can complicate genetic diagnosis and sequencing. Liaise with local genomic scientists to confirm the genetic diagnosis in these cases. Confirmatory functional testing of p47phox and p67phox expression should be sought.
Previously, contiguous gene deletions would be identified through chromosome microarray, which can be helpful in mapping out the breakpoints and genes included. Copy number variants can now be detected through whole genome sequencing.
Inheritance and genetic counselling
X-linked CGD is an X-linked recessive condition.
- X-linked recessive conditions are usually only present in males.
- Males with X-linked conditions cannot pass the variant on to their sons, but they always pass their affected X chromosome to their daughters. If the condition is recessive, their daughters will be carriers for the condition.
- Female carriers of X-linked recessive conditions have a second, working copy of the gene and are therefore usually unaffected, or affected only mildly.
- Sons of female carriers of X-linked recessive conditions have a 1-in-2 (50%) chance of being affected by the condition, and their daughters have a 1-in-2 (50%) chance of being carriers.
A family history should be taken, and parents and other potentially affected family members should be identified and screened as appropriate. Mothers are typically carriers, though de novo variants may arise.
Reproductive options are available and genetic counselling, ideally prior to conception, is recommended. Options are likely to include testing in early pregnancy or preimplantation genetic testing.
CGD types 1 to 5 are autosomal recessive conditions.
If both parents are carriers of an autosomal recessive condition, with each pregnancy there is a:
-
- 1-in-4 (25%) chance of the child inheriting both gene copies with the pathogenic variant and therefore being affected;
- 1-in-2 (50%) chance of the child inheriting one copy of the gene with the pathogenic variant and one normal copy, and therefore being a healthy carrier themselves; and
- 1-in-4 (25%) chance of the child inheriting both normal copies and being neither affected nor a carrier.
A family history should be taken, and parents and other potentially affected family members should be identified and screened as appropriate. Both parents will typically be carriers of CGD, though de novo variants may also arise. Carriers do not appear to be affected by disease.
As with X-linked CGD, reproductive options are available and genetic counselling, ideally prior to conception, is recommended.
If you are discussing genomics concepts with your patients, you may find it helpful to use the visual communication aids for genomics conversations.
Management
Management of individuals with CGD is complex and should be delivered via a multidisciplinary team in order to reduce morbidity and mortality.
Live vaccinations, including BCG, are contraindicated. Clinical management focuses on treating infections with prolonged courses of antimicrobials, and preventing infections using prophylactic antibiotics and antifungals.
Inflammatory complications may require immunomodulatory treatment and ongoing monitoring. Dermatitis and frequent skin abscesses require specialist dermatology input. Patients frequently have gastrointestinal complications that require specialist input, including biological therapies such as vedolizumab (anti-α4β7 integrin).
A curative treatment option is haematopoietic stem cell transplantation (HSCT). This involves replacing stem cells from the patient’s bone marrow with that of a compatible donor. Patients who receive allogeneic HSCT soon after birth tend to have the best outcomes with fewer complications.
Gene therapy for X-linked CGD and CGD type 1 is also in development, and may be an option in patients in whom HSCT is contraindicated.
CGD may be identified before any symptoms appear, for example through the Generation Study. Management of these individuals may differ from those presenting symptomatically.
Further information about management can be found in the resources section below.
Resources
For clinicians
- GeneReviews: Chronic granulomatous disease
- National Institute of Allergy and Infectious Diseases: Chronic granulomatous disease (CGD)
References:
- O’Donovan CJ, Tan LT, Abidin MAZ and others. ‘Diagnosis of chronic granulomatous disease: Strengths and challenges in the genomic era‘. Journal of Clinical Medicine 2024: volume 13, issue 15. DOI: 10.3390/jcm13154435
For patients
- CGD Society
- National Institute of Allergy and Infectious Diseases: Chronic granulomatous disease