ZAP70 deficiency
ZAP70 deficiency is a rare immunodeficiency condition that causes early-onset susceptibility to severe or life-threatening infections.
Overview
ZAP70 deficiency is a rare genetic condition that results in a lack of functional ZAP70, a potent intracellular signal transducer in immune cells. ZAP70 becomes phosphorylated upon TCR activation and signals to downstream immune pathways, such as mitogen-activated protein kinase (MAPK) pathway, to allow T cell proliferation, migration and differentiation, particularly CD8 T cells. Deficiency of ZAP70 causes combined immunodeficiency, which presents early in life with recurrent, persistent and severe infections.
Clinical features
ZAP70 deficiency typically results in combined immunodeficiency and a lack of functional T- and B-cell activity. In the most critical cases, patients present in infancy with severe combined immunodeficiency (SCID).
The main clinical features include:
- susceptibility to infections:
- opportunistic infections, such as Pneumocystis jirovecii pneumonia;
- recurrent upper and lower respiratory tract infections, such as cytomegalovirus pneumonitis and otitis media;
- viral infections, particularly skin warts; and
- oral candidiasis;
- autoimmunity:
- autoimmune cytopaenias; and
- autoimmune nephritis;
- immune dysregulation:
- ulcerative colitis; and
- enteropathy (presenting with chronic diarrhoea);
- malignant and non-malignant proliferation:
- predisposition to lymphoma; and
- lymphadenopathy; and
- failure to thrive.
Genomics
ZAP70 deficiency is caused by biallelic pathogenic genetic variants in the ZAP70 gene, which encodes the zeta-chain-associated protein kinase 70.
Missense, nonsense, frameshift and splice-site variants, as well as intragenic deletions, have all been reported. Pathogenic variants are distributed throughout the gene, including the N-terminal and C-terminal SH2 domains, though they are predominantly found in the kinase domain.
Null variants abolish ZAP70 protein expression with absent T cells. In these instances, patients tend to present early in life with a SCID phenotype. Biallelic missense variants interfere with phosphorylation and particularly affect the kinase domain, resulting in reduced kinase activity or instability of ZAP70 protein. Splice-site variants have also been described, which lead to aberrant messenger RNA (mRNA) processing and result in absent functional ZAP70 protein.
ZAP70 deficiency has been described all over the world, but some geographical clustering effects are seen, particularly in Middle Eastern populations.
Figure 1: T-cell activation and signal transduction via T-cell receptor/major histocompatibility complex
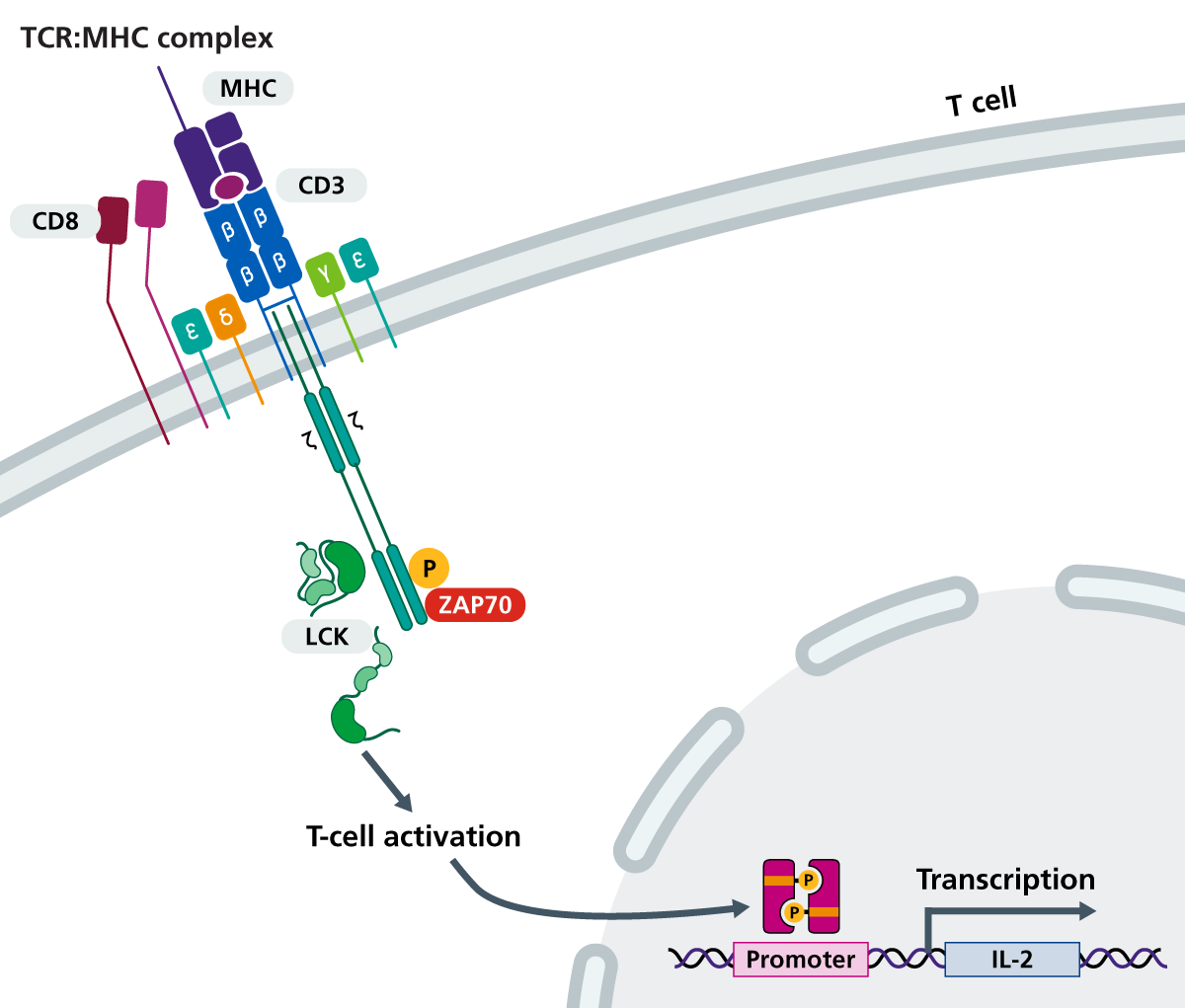
The T-cell receptor (TCR) links with major histocompatibility complex (MHC) carrying antigen. Upon assembly, the TCR associates with LCK and phosphorylates ZAP-70. ZAP-70 phosphorylates additional downstream effectors, including the adaptors Slp-76 and Lat. ZAP70 is highlighted in red. (Click to enlarge in a new tab.)
Diagnosis
ZAP70 deficiency can be diagnosed following clinical evaluation or through newborn blood spot screening using the T-cell receptor excision circles assay. Where either is suggestive of SCID, laboratory evaluation is required. Patients may have a family history of immunodeficiency.
Laboratory tests may show:
- very low CD8 T cells with near normal CD4 T-cell count (this is the main characteristic immunophenotype, and results in an elevated CD4:CD8 ratio);
- normal B and NK cell numbers (usually);
- reduced lymphocyte count in full blood count;
- hypogammaglobulinaemia and reduced antibody responses showing in immunoglobin levels (IgG levels may be reflective of maternal levels in infants less than six months old); and
- impaired response in T-cell proliferation assay.
Genomic testing confirms the diagnosis and differentiates ZAP70 deficiency from other types of primary immunodeficiency.
European Society for Immunodeficiencies diagnostic criteria is listed below.
- At least one of the following:
- at least one severe infection (requiring hospitalisation);
- one manifestation of immune dysregulation (such as autoimmunity, inflammatory bowel disease, severe eczema, lymphoproliferation and/or granuloma);
- malignancy; and
- affected family member; and
- two of the following four T-cell criteria fulfilled:
- reduced CD3, CD4 or CD8 T cells (using age-related reference values);
- reduced naive CD4 and/or CD8 T cells;
- elevated gamma-delta (γδ) T cells; and/or
- reduced proliferation to mitogen or TCR stimulation; and
- HIV excluded; and
- exclusion of a clinical diagnosis associated with combined immunodeficiency (such as defined syndromic diseases, dyskeratosis congenita or ataxia telangiectasia).
ZAP70 deficiency may be identified before any symptoms appear, for example through the Generation Study. Confirmation of the diagnosis will require referral to clinical immunology services. Please refer to the local pathway for your region for this condition.
Inheritance and genetic counselling
ZAP70 deficiency is a rare primary immunodeficiency caused by biallelic pathogenic genetic variants in the ZAP70 gene. It is inherited in an autosomal recessive pattern.
If both parents are carriers of an autosomal recessive condition, with each pregnancy there is a:
-
- 1-in-4 (25%) chance of the child inheriting both gene copies with the pathogenic variant and therefore being affected;
- 1-in-2 (50%) chance of the child inheriting one copy of the gene with the pathogenic variant and one normal copy, and therefore being a healthy carrier themselves; and
- 1-in-4 (25%) chance of the child inheriting both normal copies and being neither affected nor a carrier.
A family history should be taken, and parents and other potentially affected family members should be identified and screened as appropriate. Note that de novo variants may also arise, and that carriers do not appear to be affected by disease.
If you are discussing genomics concepts with your patients, you may find it helpful to use the visual communication aids for genomics conversations.
Management
ZAP70 deficiency is a serious genetic condition that can be fatal without early treatment. Generally, immediate supportive care is required for patients presenting with infections. This includes:
- no live vaccinations;
- aggressive treatment of infections, including with anti-fungal and anti-viral agents;
- infection prevention;
- immunoglobulin replacement therapy;
- nutritional support; and
- irradiated, cytomegalovirus negative blood products, if required.
The primary curative treatment is haematopoietic stem cell transplantation (HSCT). This involves replacing stem cells from the patient’s bone marrow with that of a compatible donor. Patients who receive allogeneic HSCT soon after birth tend to have the best outcomes with fewer complications.
ZAP70 deficiency may be identified before any symptoms appear, for example through the Generation Study. Management of these individuals may differ from those presenting symptomatically.
Resources
For clinicians
- European Society for Immunodeficiencies: Diagnosis criteria
- Jeffrey Modell Foundation: 10 warning signs of primary immunodeficiency
- OMIM: 269840 Immunodeficiency 48
References:
- Cuvelier GDE, Rubin TS, Wall DA and others. ‘Long-term outcomes of hematopoietic stem cell transplantation for ZAP70 deficiency‘. Journal of Clinical Immunology 2016: volume 36, issue 7, pages 713–724. DOI: 10.1007/s10875-016-0316-z
- Sharifinejad N, Jamee M, Zaki-Dizaji M and others. ‘Clinical, immunological, and genetic features in 49 patients with ZAP-70 deficiency: A systematic review‘. Frontiers in Immunology 2020: volume 11, page 831. DOI: 10.3389/fimmu.2020.00831
For patients
- Great Ormond Street Hospital for Children NHS Foundation Trust: Combined immunodeficiency (CID) in children
- Immunodeficiency UK: Combined immunodeficiency in children