X-linked agammaglobulinaemia
X-linked agammaglobulinaemia is a primary B-cell immunodeficiency characterised by susceptibility to infections, low/absent serum immunoglobulins and low/absent B cells. It is caused by pathogenic genetic variants in the BTK gene, which codes for a critical tyrosine kinase in B-cell development.
Overview
X-linked agammaglobulinaemia (XLA) is a humoral immunodeficiency with reduced or absent antibody production and low/absent B cells. Affected boys typically present in the first two years with recurrent bacterial infections, such as otitis media and pneumonia. Conjunctivitis, sinusitis, diarrhoea and skin infections are also seen frequently. The first presentation may be with a severe, life-threatening infection.
Clinical features
Clinical features of XLA include:
- increased susceptibility to extracellular bacterial infections, typically lower respiratory tract infections, sinusitis and otitis media;
- increased susceptibility to mycoplasma and giardia infection;
- severe or prolonged enteroviral infections;
- early-onset neutropenia, which typically improves with time;
- bronchiectasis;
- chronic diarrhoea and colitis; and
- rarely, autoimmune manifestations, such as arthritis and hypothyroidism.
Affected patients typically present in the first year of life, once maternal transplacental IgG have declined – usually six to eight weeks after birth.
The first presentation in affected individuals may be a severe, life-threatening infection, such as pneumonia, empyema, meningitis, sepsis, cellulitis or septic arthritis.
Female carriers of XLA are typically unaffected, though a small number have been reported to be symptomatic as a result of skewed X-inactivation.
Genomics
XLA is an X-linked recessive condition caused by pathogenic variants in the BTK gene (see Figure 1). Affected individuals are almost all male; either they have inherited the variant from their mother, who will be a carrier for the condition, or the variant has arisen de novo.
A variety of causative variants have been identified, including nonsense, missense, splice site, intronic, large-scale deletions and promoter deletions. Some missense and splice site variants are associated with a milder presentation. Some larger deletions involve other genes, for instance the closely linked gene TIMM8A, which causes hearing impairment.
There have been occasional cases of girls with XLA due to skewed inactivation of the X chromosome (in which the X chromosome carrying the standard BTK variant is preferentially silenced).
In males, molecular investigation of suspected agammaglobulinaemia should start with genetic analysis of BTK, variants in which account for 85% of agammaglobulinaemia. In females, or males in whom XLA has been excluded, autosomal recessive and dominant causes of agammaglobulinaemia should be considered.
Figure 1: Errors in B-cell development
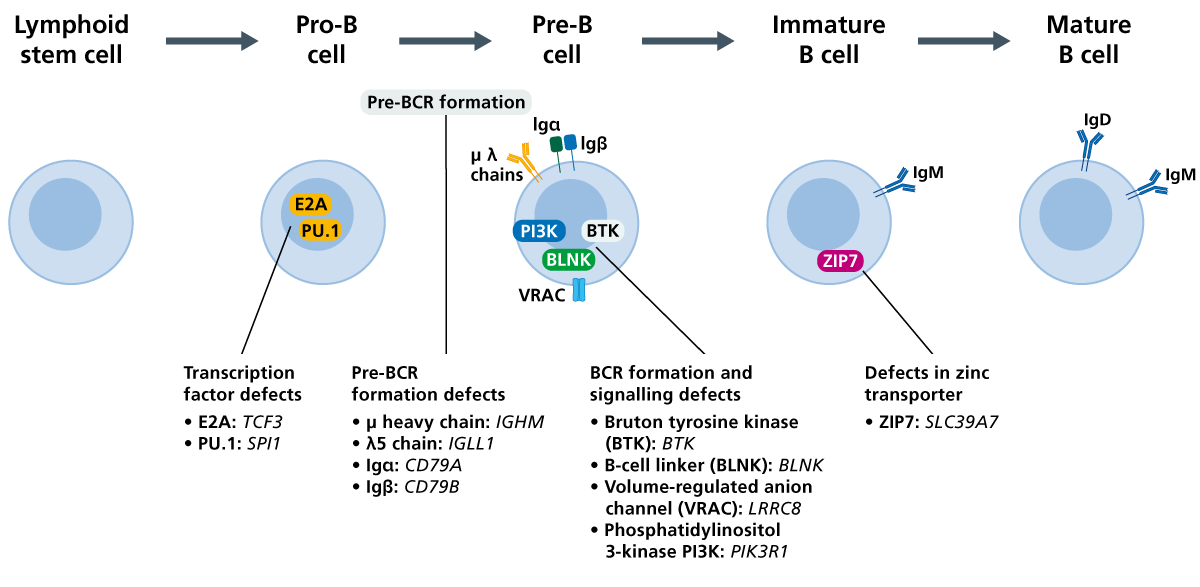
Pathogenic variants in the BTK gene, essential for B-cell development, are the cause of cause X-linked agammaglobulinaemia. (Click to enlarge in a new tab.)
Diagnosis
According to the European Society For Immunodeficiencies diagnostic criteria, a diagnosis of XLA should be considered on identifying the following:
- male patient with less than 2% CD19+ B cells;
- onset of recurrent bacterial infections in the first five years of life;
- serum IgG, IgM and IgA < 2 SD for normal for age;
- absent isohemagglutinins and /or poor response to vaccines; and
- other causes of hypogammaglobulinaemia have been excluded.
Confirmation of the diagnosis in a male patient requires the finding of less than 2% CD19+ B cells with at least one of the following:
- pathogenic variant in BTK;
- absent BTK mRNA on northern blot analysis of neutrophils or monocytes;
- absent BTK protein in monocytes or platelets; and/or
- maternal cousins, uncles or nephews with less than 2% CD19+ B cells.
For information about genomic testing, see ‘Child with agammaglobulinaemia with absent BTK expression‘.
XLA may be identified before any symptoms appear, for example through the Generation Study. Confirmation of the diagnosis will require referral to clinical immunology. Please refer to the local pathway for your region for this condition.
Inheritance and genetic counselling
XLA is an X-linked recessive condition.
- X-linked recessive conditions are usually only present in males.
- Males with X-linked conditions cannot pass the variant on to their sons, but they always pass their affected X chromosome to their daughters. If the condition is recessive, their daughters will be carriers for the condition.
- Female carriers of X-linked recessive conditions have a second, working copy of the gene and are therefore usually unaffected, or affected only mildly.
- Sons of female carriers of X-linked recessive conditions have a 1-in-2 (50%) chance of being affected by the condition, and their daughters have a 1-in-2 (50%) chance of being carriers.
A family history should be taken, and screening and/or testing for the familial BTK variant should be offered to at-risk relatives. Reproductive options are available and genetic counselling is recommended. Note that de novo variants account for 15%–20% of all cases of XLA.
If you are discussing genomics concepts with your patients, you may find it helpful to use the visual communication aids for genomics conversations.
Management
The mainstay of treatment for XLA is immunoglobulin replacement therapy. This is available in many different preparations, and can be administered intravenously or subcutaneously. Prophylactic antibiotics may also be used to prevent infections, and prompt treatment with antibiotics for a longer course is required when the patient develops an infection. Long-term monitoring for infectious complications is also required.
XLA may be identified before any symptoms appear, for example through the Generation Study. Management of these individuals may differ from those presenting symptomatically.
Further information about management can be found in the resources section below.
Resources
For clinicians
- European Society for Immunodeficiencies: Diagnosis criteria
- GeneReviews: X-linked agammaglobulinemia
- NHS England: Clinical Commissioning Policy for the use of therapeutic immunoglobin (Ig) England (2025) (PDF, 37 pages)
References:
- Shillitoe B and Gennery A. ‘X-linked agammaglobulinaemia: Outcomes in the modern era‘. Clinical Immunology 2017: volume 183, pages 54–62. DOI: 10.1016/j.clim.2017.07.008
- Takada H, Kanegane H, Nomura A and others. ‘Female agammaglobulinemia due to the Bruton tyrosine kinase deficiency caused by extremely skewed X-chromosome inactivation‘. Blood 2004: volume 103, issue 1, pages 185–187. DOI: 10.1182/blood-2003-06-1964#