STAT1 deficiency
STAT1 deficiency is a rare genetic condition caused by defects in a critical immune transcription factor called STAT1, which lead to severe mycobacterial and viral infections.
Overview
STAT1 deficiency is a very rare primary immunodeficiency condition characterised by early onset severe mycobacterial and/or viral infections. Complete deficiency of the STAT1 transcription factor abolishes cellular responses to interferon signalling, particularly IFN-γ (see figure 1).
Figure 1: The IL-12/23-IFN-γ pathway in T cells and phagocytes
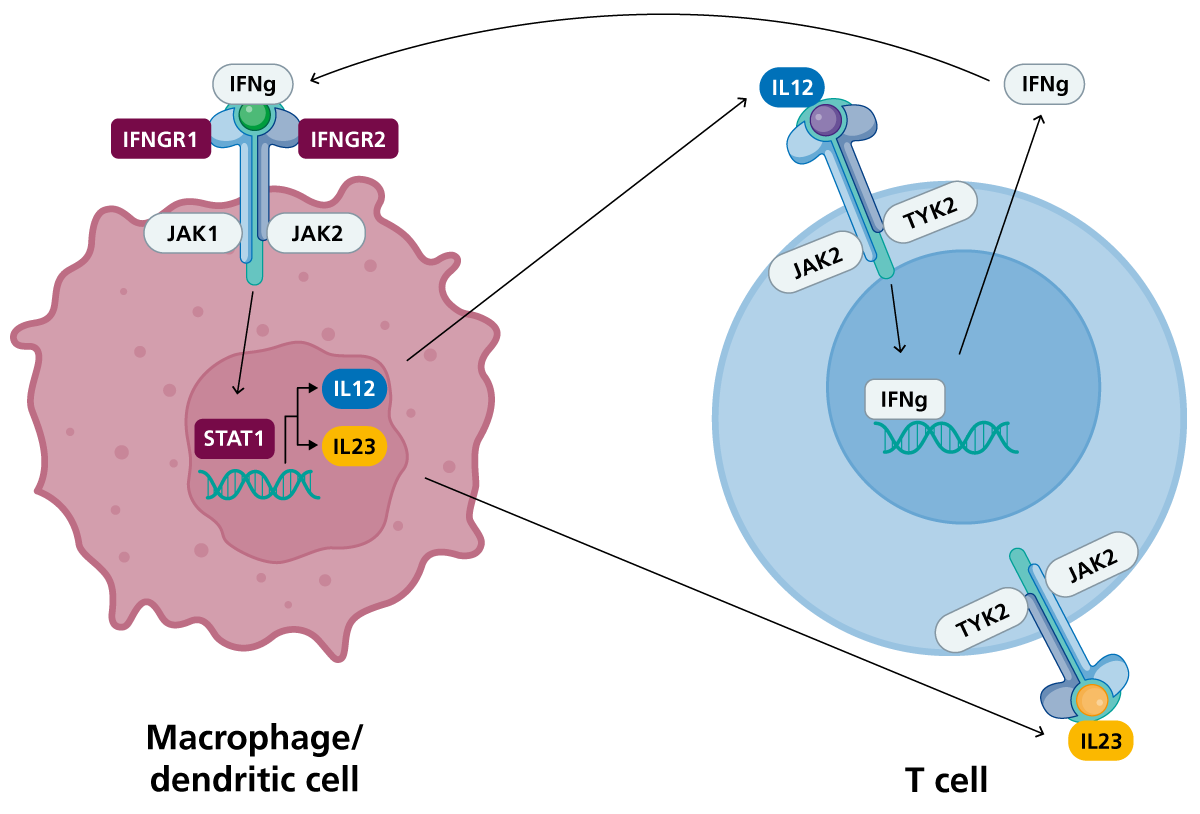
Upon recognition of the pathogen, phagocytes (or dendritic cells) secrete IL-12 and IL-23, which bind to their receptors on T cells, inducing the production of IFN-γ by STAT1 transcription factor binding. IFN-γ can bind to IFN-γ receptor on phagocytes to promote phagocyte-mediated killing of the pathogen. (Click to enlarge in a new tab.)
Clinical features
STAT1 deficiency causes a syndrome called Mendelian susceptibility to mycobacterial disease (MSMD). Infections typically present in early childhood.
- Affected patients will have increased susceptibility to typical and atypical mycobacterial and viral infections.
- M. tuberculosis infection is often severe and prolonged, and may be disseminated.
- The Bacillus Calmette-Guérin (BCG) vaccine may develop into disseminated disease in affected vaccinated patients.
- Non-tuberculous mycobacterial (NTM) infections can occur, particularly affecting the respiratory tract.
- Affected patients will have increased susceptibility to opportunistic infections and fungal infections.
Other variable clinical features include:
- failure to thrive;
- granulomatous inflammation;
- hepatosplenomegaly; and
- chronic fever.
Complete STAT1 deficiency presents at an early age with severe disease and high mortality.
Partial STAT1 loss-of-function disease has also been observed. This has a milder phenotype and tends to present later with MSMD or recurrent bacterial and/or viral infections. Infections may be more amenable to treatment in this group.
Genomics
Pathogenic constitutional (germline) missense, nonsense, frameshift and splice site variants in STAT1 have been reported to cause immunodeficiency or immune dysregulation.
Autosomal recessive STAT1 deficiency is caused by biallelic (homozygous/compound heterozygous) pathogenic variants in the STAT1 gene. These are typically loss-of-function (often null) variants, which lead to complete absence of STAT1 function.
Autosomal dominant STAT1 deficiency has also been described. This has a similar phenotype but is caused by monoallelic (heterozygous) loss-of-function STAT1 variants or STAT1 variants that confer a dominant negative effect and interfere with wild-type STAT1 function.
The clinical phenotype and severity depend on the specific variant and its impact on STAT1 expression and function.
Diagnosis
STAT1 deficiency may be diagnosed following clinical evaluation of recurrent or severe mycobacterial or viral infections. Patients may have a family history of early onset immunodeficiency.
In laboratory tests:
- STAT1 phosphorylation following IFN-γ stimulation is reduced/absent compared to control, suggesting defective IFN-γ signalling;
- IFN-γ receptor expression is normal in complete deficiency, although may be present in partial forms; and
- IFN-γ cytokine levels are usually lower than controls.
Genomic testing confirms the diagnosis and differentiates STAT1 deficiency with other forms of primary immunodeficiency known to cause MSMD.
The European Society for Immunodeficiencies diagnostic criteria for inborn errors of mycobacterial susceptibility are:
- infections caused by weakly virulent mycobacteria, such as BCG vaccines and environmental mycobacteria, tuberculosis, salmonellosis, candidiasis, other intramacrophagic bacteria, fungi or parasites; and
- altered IFN-γ mediated immunity tests; or
- altered IL-12 mediated immunity tests; and
- no IFN-γ auto-antibodies.
For information about testing, see ‘Infant or child with severe, recurrent, persistent and/or unusual infections‘.
STAT1 deficiency may be identified before any symptoms appear, for example through the Generation Study. Confirmation of the diagnosis will require referral to clinical immunology services. Please refer to the local pathway for your region for this condition.
Inheritance and genetic counselling
STAT1 deficiency is caused by biallelic pathogenic variants in the STAT1 gene. Complete deficiency shows autosomal recessive inheritance. Autosomal dominant inheritance is also recognised with partial STAT1 deficiency.
A family history should be taken, and parents and other potentially affected family members should be identified and screened as appropriate. Note that de-novo variants may also arise, and that heterozygous carriers may have symptoms of immunodeficiency or immune dysregulation.
- If both parents are carriers of an autosomal recessive condition, with each pregnancy there is a:
- 1-in-4 (25%) chance of a child inheriting both gene copies with the pathogenic variant and therefore being affected;
- 1-in-2 (50%) chance of a child inheriting one copy of the gene with the pathogenic variant and one normal copy, and therefore being a healthy carrier themselves; and
- 1-in-4 (25%) chance of a child inheriting both normal copies and being neither affected nor a carrier.
- Individuals affected by an autosomal dominant condition have one working copy of the gene, and one with a pathogenic variant.
- The chance of a child inheriting the gene with the variant from an affected parent is 1 in 2 (50%).
- Incomplete penetrance can occur (that is, not everyone who has the variant develops the disease).
- If you are discussing genomics concepts with your patients, you may find it helpful to use the visual communication aids for genomics conversations.
Management
Complete STAT1 deficiency is a serious life-threatening genetic condition that can be fatal without early and aggressive treatment. Generally, immediate supportive care is required for patients presenting with infections. This includes:
- no live vaccinations, particularly the BCG vaccination;
- aggressive treatment of infections with antimycobacterial and/or antiviral agents, typically using combinations of different agents for several months; and
- infection prevention with prophylactic antibiotics and regular infection surveillance.
The primary curative treatment is haematopoietic stem cell transplantation (HSCT). This involves replacing stem cells from the patient’s bone marrow with that of a compatible donor. Patients who receive allogeneic HSCT soon after birth tend to have the best outcomes with fewer complications.
Patients with partial deficiency (autosomal dominant, dominant negative disease) may not require HSCT, particularly as infection may be more responsive to anti-mycobacterial medication and infection risk potentially decreases into adulthood.
STAT1 deficiency may be identified before any symptoms appear, for example through the Generation Study. Management of these individuals may differ from those presenting symptomatically.
Resources
For clinicians
References:
- Dupuis S, Dargemont C, Fieschi C and others. ‘Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation‘. Science 2001: volume 293, issue 5,528, pages 300–303. DOI: 10.1126/science.1061154
- Dupuis S, Jouanguy E, Al-Hajjar S and others. ‘Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency‘. Nature Genetics 2003: volume 33, issue 3, pages 388–391. DOI: 10.1038/ng1097
For patients
- Immune Deficiency Foundation: Innate immune disorders
- Immunodeficiency UK