Palmoplantar epidermal differentiation disorders (palmoplantar keratoderma)
Palmoplantar epidermal differentiation disorders encompass a group of disorders that cause excessive skin thickening, primarily affecting the palms and soles. They can present in early childhood or later in life, depending on the type. Some forms are hereditary, caused by pathogenic variants in keratin-related genes, and follow autosomal dominant or recessive inheritance patterns.
Overview
Palmoplantar epidermal differentiation disorders (pEDDs) are a group of genetic conditions characterised by abnormal thickening of the skin on the palms and soles. Pain arising from plantar thickened, fissured skin may limit the ability for affected patients to walk. Similarly, changes in palmar skin can significantly affect activities of daily living and work. Testing for pEDDs should be reserved for cases where other non-genomic clinical diagnoses that cause palmoplantar skin thickening, such as fungal infections, have been excluded.
Clinical features
Clinical features of pEDDs include:
- thickened, scaly or waxy skin on palms and soles;
- pain and difficulty walking in severe cases;
- redness or yellow discoloration of affected areas;
- increased susceptibility to fungal infections; and
- nail thickening and discoloration.
Genomics
pEDDs are often linked to pathogenic variants in genes encoding keratin proteins, such as KRT1 and KRT9. These pathogenic variants disrupt the structural integrity of the skin, leading to thickening and scaling. Genomic testing focuses on identifying these pathogenic variants to confirm diagnosis and guide management.
Some causative genes are also associated with broader health implications. For instance, KRT6A, KRT6B, KRT6C, KRT16 and KRT17 are responsible for a group of pEDDs previously labelled pachyonychia congenita (figures 1 and 2). In some of these patients, additional features such as sebaceous cysts and hidradenitis suppuritiva may be seen.
The naming system for pEDD has recently changed from palmoplantar keratodermas (PPK) and will be underpinned by genomic testing. The new names have two parts consisting of:
- the gene that is affected; and
- the type of EDD – which, in the case of the palmoplantar keratodermas, will be pEDD (palmoplantar epidermal differentiation disorders).
For example, pachyonychia congenita caused by KRT16 pathogenic variants is now “KRT16-pEDD”.
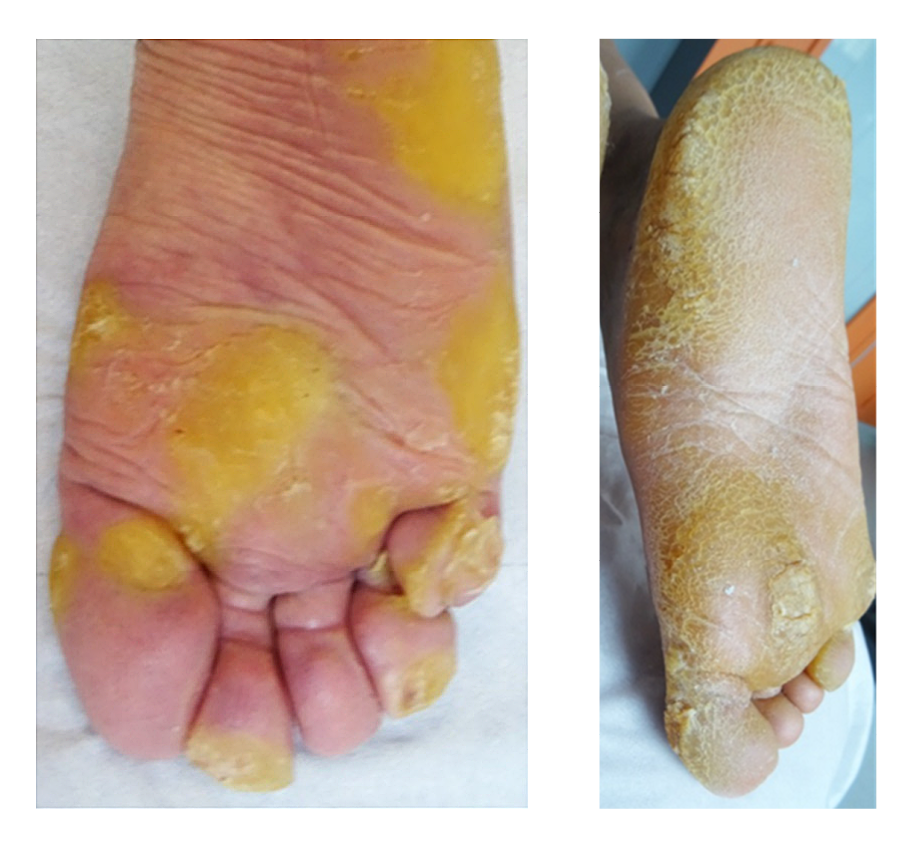
Figure 1 (left): Plantar hyperkeratosis in a patient with KRT16-pEDD
Figure 2 (right): Plantar hyperkeratosis in a patient with KRT6A-pEDD
Images reproduced from ‘Treatment of Painful Palmoplantar Keratoderma Related to Pachyonychia Congenita Using EGFR Inhibitors‘, under MDPI’s open access licence.
Diagnosis
pEDDs can be diagnosed by:
- clinical examination of skin thickening and distribution;
- histological analysis of affected skin; and
- in some cases, genomic testing with a panel in the National Genomic Test Directory (R166 Palmoplantar keratodermas) to identify pathogenic variants in causative genes. For more information about testing, see Patient with a personal and family history of thickened palmoplantar skin.
Inheritance and genomic counselling
Hereditary forms of pEDD can follow autosomal dominant or recessive inheritance patterns. Autosomal dominant forms are more common. Genomic counselling is essential to discuss inheritance risks and implications for family members.
For autosomal dominant inheritance patterns:
- Individuals affected by an autosomal dominant condition have one working copy of the gene, and one with a pathogenic variant.
- The chance of a child inheriting the gene with the variant from an affected parent is 1-in-2 (50%).
For autosomal recessive inheritance patterns:
- If both parents are carriers of an autosomal recessive condition, with each pregnancy there is a:
- 1-in-4 (25%) chance of a child inheriting both gene copies with the pathogenic variant and therefore being affected;
- 1-in-2 (50%) chance of a child inheriting one copy of the gene with the pathogenic variant and one normal copy, and therefore being a healthy carrier themselves; and
- 1-in-4 (25%) chance of a child inheriting both normal copies and being neither affected nor a carrier.
Incomplete penetrance can occur when not everyone who has the variant develops the disease.
Reproductive options are available if patients are concerned about having an affected child. This can include testing in early pregnancy, or potentially preimplantation genetic diagnosis.
Management
Management involves symptomatic treatment, such as moisturising and addressing infections. Good foot care and footwear are important. Secondary fungal and bacterial infections that require treatment are common.
For some, genotyped patient trials involving repurposed treatments such as EGFR inhibitors are available. Genetic therapies are not yet available, but ongoing research may provide future options.
Some genetic causes will have broader health and management implications.
Resources
For clinicians
- DermNet: Palmoplantar keratoderma
References:
- Greco C, Ponsen A-C, Leclerc-Mercier S and others. ‘Treatment of Painful Palmoplantar Keratoderma Related to Pachyonychia Congenita Using EGFR Inhibitors‘. MDPI Biomedicines 2022: volume 10, issue 4, page 841. DOI: 10.3390/biomedicines10040841
- Hernandez-Martin A, Paller AS, Sprecher E and others. ‘A proposal for a new pathogenesis-guided classification for inherited epidermal differentiation disorders‘. British Journal of Dermatology 2025: volume 193, issue 3, pages 544–548. DOI: 10.1093/bjd/ljaf065
- Sprecher E, Ishida-Yamamoto A, Schwartz J and others. ‘Palmoplantar epidermal differentiation disorders: a new classification toward pathogenesis-based therapy‘. British Journal of Dermatology 2025: volume 193, issue 3, pages 364–380. DOI: 10.1093/bjd/ljaf054
For patients
- British Association of Dermatologists: Palmoplantar keratoderma