IL2RG-associated X-linked severe combined immunodeficiency
IL2RG-associated X-linked severe combined immunodeficiency (SCID), also called common gamma chain (γc) SCID or X-SCID, is a rare inherited immunodeficiency condition characterised by severely reduced T- and NK-cell function. It accounts for nearly 50% of SCID cases.
Overview
IL2RG-associated X-linked severe combined immunodeficiency (SCID), or X-SCID, is caused by pathogenic genetic variants in the common gamma chain (γc) of the IL-2 receptor. This is a shared component of multiple critical immune pathways and causes defects in T- and NK-cell development. Patients present early in infancy with severe, persistent and opportunistic infections. Early diagnosis permits better treatment outcomes, primarily through haematopoietic stem cell transplantation. Untreated, the condition is typically fatal within 12 months.
Clinical features
Patients present with features of SCID, a life-threatening condition resulting from severely impaired T-cell function. IL2RG-associated SCID symptoms are generally indistinguishable from other forms of SCID, although male sex makes IL2RG-associated SCID more likely. Patients may be asymptomatic at birth.
Typical infections include severe pneumonia or sepsis due to opportunistic pathogens such as Pneumocystis jiroveci, Pseudomonas sp, Candida sp and cytomegalovirus. Infections tend to be persistent, invasive and life-threatening, despite conventional treatment. Vaccine-strain infection following administration of live vaccinations, such as BCG or rotavirus, may also occur.
Features of graft-versus-host-disease can occur due to lymphocytic infiltration, resulting in:
- maculopapular skin rash;
- chronic diarrhoea; and/or
- jaundice or hepatomegaly.
Other features can include:
- failure to thrive;
- oral thrush; and
- absent tonsils or peripheral lymph nodes.
As transplacental immunity wanes, infants usually develop infections by six months of age. Persistence of symptomatic infection following administration of live vaccinations in the first few months of life may occur. Some infants develop an erythematous rash associated with spontaneous graft-versus-host disease from birth.
Hypomorphic variants in IL2RG may allow partial kinase activity, which would lead to an atypical presentation of X-SCID, often with milder symptoms and later onset. Autoimmune and lymphoproliferative manifestations occur more often in hypomorphic X-SCID.
Genomics
IL2RG encodes the gamma chain (γc) of the IL-2 receptor, which is found in multiple cytokine pathways. The disruption in signalling causes absent or reduced IL-7 signalling, impacting T-cell development and IL-15 signalling, and impairing NK-cell development.
Many hundreds of different pathogenic variants have been described in IL2RG-associated X-SCID across all populations, and there is recognised genotype-phenotype correlation, as outlined below.
- Classic X-SCID with profound immunodeficiency typically results from complete loss-of-function (null) variants, such as nonsense, frameshift and large deletions.
- Hypomorphic X-SCID, in which there is some residual T-cell function and a milder phenotype, can be seen with some missense and splice-site variants.
- p.Arg222Cys-related X-SCID typically features near normal numbers of T cells and NK cells, though all reported individuals with this variant have had opportunistic infections within the first year of life. Affected individuals present along a spectrum of classic to more atypical hypomorphic X-SCID.
Figure 1: IL-7 signalling via common gamma chain (γc) and IL-7 receptor
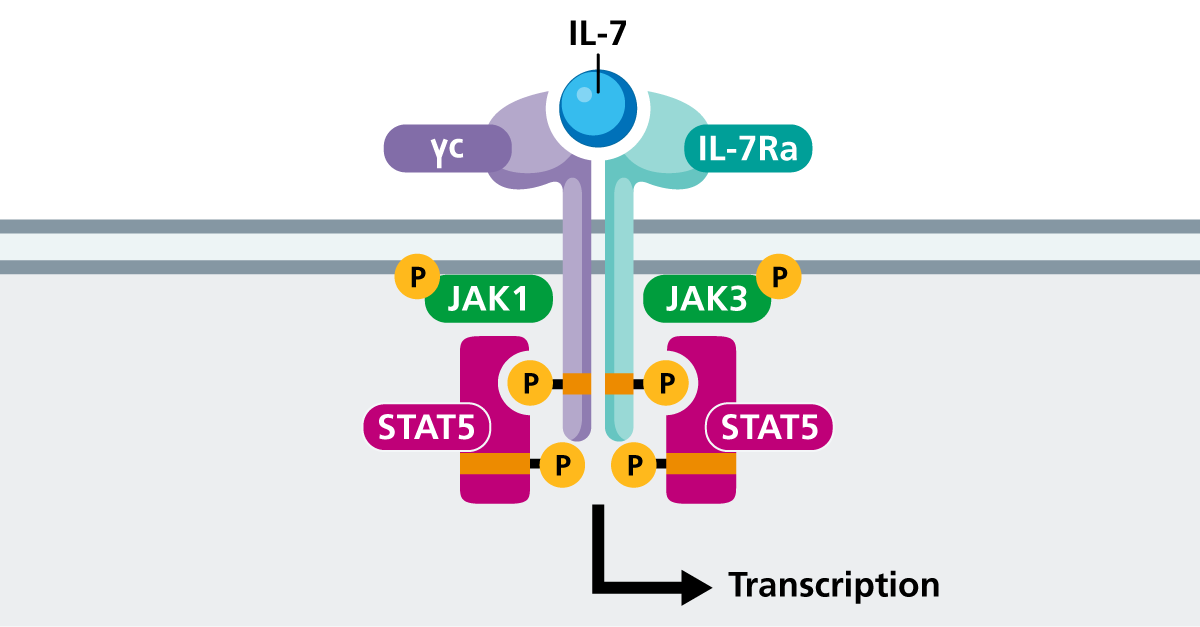
(Click to enlarge in a new tab.)
Diagnosis
IL2RG-associated SCID can be diagnosed following clinical evaluation or through the newborn blood spot screening programme using the T-cell receptor excision circles assay. If either is suggestive of SCID, prompt laboratory evaluation is required. Patients may have a family history of immunodeficiency – for example, a history of affected male relatives.
In laboratory tests:
- full blood count may show reduced lymphocyte count;
- lymphocyte subset testing will show absent or very low levels of T cells and absent or very low levels of NK cells, with normal or near normal B cell count (deficiency of naive T cells is characteristic of SCID);
- immunoglobulin levels will show reduced antibody responses (IgG levels may be reflective of maternal levels in infants less than six months old); and
- T-cell proliferation assay will show impaired response.
Genomic testing confirms the diagnosis and differentiates IL2RG X-SCID from other types of SCID.
The European Society for Immunodeficiencies diagnostic criteria for SCID is outlined below.
- At least one of the following:
- invasive bacterial, viral or fungal/opportunistic infection;
- persistent diarrhoea and failure to thrive; and/or
- affected family member; and
- manifestation in the first year of life; and
- HIV excluded; and
- two of the following four T-cell criteria are fulfilled:
- low or absent CD3, CD4 or CD8 T cells;
- reduced naive CD4 and/or CD8 T cells;
- elevated gamma delta (γδ) T cells; and/or
- reduced or absent proliferation to mitogen or T-cell receptor stimulation.
For information about testing, see ‘Infant with severe combined immunodeficiency with features of gamma chain deficiency‘.
IL2RG-associated X-SCID may be identified before any symptoms appear; for example, through the Generation Study. Confirmation of the diagnosis will require referral to clinical immunology services. Please refer to the local pathway for your region for this condition.
Inheritance and genetic counselling
IL2RG-associated X-SCID is caused by pathogenic genetic variants in the IL2RG gene, which lies on the X chromosome. It is inherited in an X-linked recessive pattern and accounts for 40%–50% of SCID cases.
- X-linked recessive conditions are usually only present in males.
- Males with X-linked conditions cannot pass the variant on to their sons, but they always pass their affected X chromosome to their daughters. If the condition is recessive, their daughters will be carriers for the condition.
- Female carriers of X-linked recessive conditions have a second, working copy of the gene and are therefore usually unaffected, or affected only mildly.
- Sons of female carriers of X-linked recessive conditions have a 1-in-2 (50%) chance of being affected by the condition, and their daughters have a 1-in-2 (50%) chance of being carriers.
A family history should be taken, and parents and other potentially affected family members should be identified and screened as appropriate. Mothers are typically asymptomatic carriers, and de novo variants commonly arise.
Reproductive options are available and genetic counselling, ideally prior to conception, is recommended. Options are likely to include testing in early pregnancy or preimplantation genetic testing.
If you are discussing genomics concepts with your patients, you may find it helpful to use the visual communication aids for genomics conversations.
Management
X-SCID is a fatal condition unless definitive treatment to reconstitute the immune system is undertaken. Immediate management principles involve treatment and prevention of infections. This includes:
- no live vaccinations;
- aggressive treatment of infections, including with anti-fungal and anti-viral agents;
- isolation from exposure to pathogens;
- immunoglobulin replacement;
- parenteral nutrition; and
- irradiated, cytomegalovirus-negative blood products, if required.
The primary curative treatment is haematopoietic stem cell transplantation (HSCT). This involves replacing stem cells from the patient’s bone marrow with that of a compatible donor. Patients who receive allogeneic HSCT soon after birth tend to have the best outcomes with fewer complications.
Another potentially curative treatment is gene therapy, which involves patient stem cells being corrected in vitro using retroviral gene transfer and then re-infused back into the patient.
IL2RG-associated X-SCID may be identified before any symptoms appear; for example, through the Generation Study. Management of these individuals may differ from those presenting symptomatically.
Resources
- European Society for Immunodeficiencies: Diagnosis criteria
- GeneReviews: X-linked severe combined immunodeficiency
- National Institutes of Health: Genetic testing registry: JAK3
- OMIM: 300400 Severe combined immunodeficiency, X-linked
References:
- Jiang C, He Y, Chen X and others. ‘X-linked severe combined immunodeficiency complicated by disseminated bacillus Calmette-Guérin disease caused by a novel pathogenic mutation in exon 3 of the IL2RG gene: a case report and literature review‘. Frontiers Immunology 2024: volume 15. DOI: 10.3389/fimmu.2024.1453046
For patients
- Great Ormond Hospital for Children NHS Foundation Trust: Severe combined immunodeficiency (SCID)
- Immunodeficiency UK: X-linked severe combined immunodeficiency