Hereditary angioedema
Hereditary angioedema is the name of a group of rare genetic conditions that cause localised non-urticarial swelling of skin or mucosa. They commonly affect the extremities, face and abdomen, and can be life-threatening when affecting the larynx. Swellings are recurrent and can be unpredictable.
Overview
Hereditary angioedema types 1 and 2 account for the majority of cases and are caused by heterozygous genetic variants in the SERPING1 gene, causing deficiency in either esterase inhibitor levels or function. Rarer genetic involvement includes F12, PLG, ANGPT1, KNG1, MYOF and HS3ST6.
Hereditary angioedema typically presents in childhood and is characterised by:
- recurrent episodes of angioedema;
- recurrent abdominal pain, lasting one to three days;
- lack of response to anti-histaminergic medication; and
- being triggered by physical injury, surgical procedures, stress, ACE inhibitors and/or oestrogen.
Clinical features
The main feature of hereditary angioedema is cutaneous or submucosal swelling due to bradykinin-mediated increased vascular permeability. This can affect any region but is most commonly seen in:
- extremities;
- gastrointestinal tract;
- face; and
- upper airway.
In addition, the affected skin is non-pruritic and swelling is non-pitting. Patients may have a prodromal serpiginous erythematous rash. Episodes of angioedema are not responsive to the following agents, which can help distinguish it from more common causes of angioedema:
- antihistamines;
- non-steroidal anti-inflammatory drugs (NSAIDs); and
- adrenaline.
Drug-related angioedema should also be excluded prior to diagnosis.
Genomics
Hereditary angioedema types 1 and 2
- Caused by heterozygous pathogenic genetic variants in the SERPING1 gene.
- In type 1 (85% of CI esterase inhibitor deficiency):
- In type 2 (low C1 inhibitor functioning):
- variants tend to be at or near the active site on the reactive mobile loop;
- mutant C1 inhibitor protein is secreted but is dysfunctional; and
- C1 esterase inhibitor levels are normal.
- A family history is often present, but up to 20% of pathogenic variants in SERPING1 arise de novo.
Hereditary angioedema with normal C1 esterase inhibitor levels and function
This condition is described in rare cases, with pathogenic variants in the following genes:
- F12:
- typically missense variants in exon 9, where all known pathogenic variants are located;
- most commonly at position 309 (historically numbered as 328); and
- variants lead to a gain-of-function in factor XII;
- PLG:
- the most common variant is in exon 9 p.Lys330Glu (K330E);
- similar to F12, has a gain-of-function mechanism;
- ANGPT1;
- KNG1;
- MYOF; and
- HS3ST6.
Diagnosis
Hereditary angioedema should be distinguished from histaminergic angioedema (IgE mediated), acquired angioedema (autoimmune or paraprotein-related) and drug-related angioedema.
If there is a suspicion of hereditary angioedema, initial investigations include:
- C1 inhibitor function;
- C1 inhibitor level; and
- C4 level.
Where there are low levels of all three, hereditary angioedema type 1 is likely. The tests may need to be repeated, as in complement testing sample degradation can occur and can impact accurate reporting.
Where C1 inhibitor function is within normal levels and both C1 inhibitor level and C4 levels are low, hereditary angioedema type 2 is likely.
In cases in which C1 inhibitor function and level is within normal range, F12 genomic testing should be sought.
Please see diagnostic criteria and consensus guidelines by international WAO/EACCI for hereditary angioedema (2021).
For more information about diagnostic criteria and consensus guidelines, see the references section below. For information about testing, see ‘Child with recurrent swellings‘. Note that, at present, there is no testing pathway for rare causes of hereditary angioedema, other than SERPING1 and F12 testing.
Inheritance and genetic counselling
Hereditary angioedema types 1 and 2 arise in about 1 in 50,000 people. Pathogenic genetic variants in SERPING1 and F12 show autosomal dominant inheritance. A family history should be taken, and parents and other potentially affected family members should be identified and screened as appropriate. De novo variants arise in about 20% of patients.
- Individuals affected by an autosomal dominant condition have one working copy of the gene, and one with a pathogenic variant.
- The chance of a child inheriting the gene with the variant from an affected parent is 1 in 2 (50%).
- Incomplete penetrance can occur (that is, not everyone who has the variant develops the disease). It is particularly reported with F12 variants.
If you are discussing genomics concepts with your patients, you may find it helpful to use the visual communication aids for genomics conversations.
Management
There are two main goals for the management of hereditary angioedema:
- control of acute swellings; and
- prevention of recurrent swellings.
Treatment of acute swellings, which can affect the upper airway, should be carried out promptly with either intravenous C1 inhibitor or icatibant (see figure 1).
To achieve long-term prevention of swellings, NICE recommends one of three agents, depending on clinical criteria and availability:
- C1 esterase inhibitor, plasma-derived or recombinant;
- lanadelumab, monoclonal antibody against plasma kallikrein; or
- berotralstat, plasma kallikrein inhibitor.
Further information about management can be found in the resources section below.
Figure 1: Bradykinin-mediated angioedema and treatment
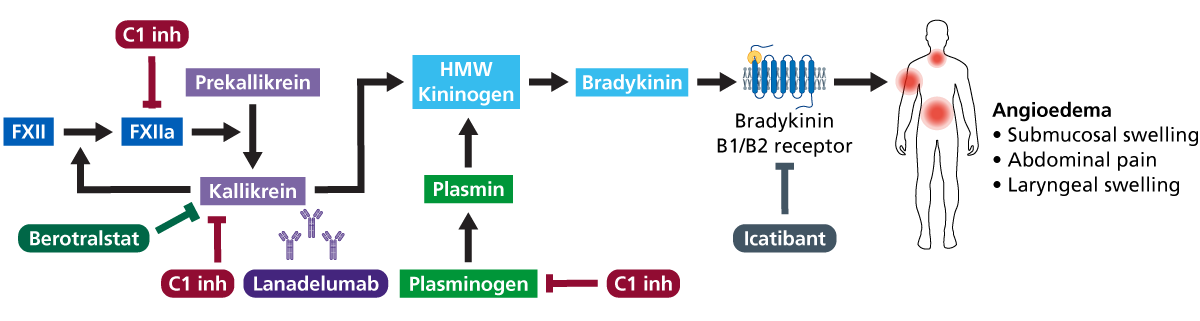
The bradykinin pathway: hereditary angioedema conditions all lie within the bradykinin-forming contact system and cause unregulated production of bradykinin. (Click to enlarge in a new tab.)
Resources
For clinicians
- British Society of Immunology: BiteSized immunology: Immune dysfunction: Hereditary angioedema
- NICE: Berotralstat for preventing recurrent attacks of hereditary angioedema
- NICE: Icatibant
- NICE: Lanadelumab for preventing recurrent attacks of hereditary angioedema
- NHS England: Clinical Commissioning Policy: Plasma-derived C1-esterase inhibitor for prophylactic treatment of hereditary angioedema (HAE) types I and II (PDF, 21 pages)
- StatPearls: Hereditary angioedema
References:
- Busse PJ and Christiansen SC. ‘Hereditary angioedema‘. The New England Journal of Medicine 2020: volume 382, issue 12, pages 1,136–1,148. DOI: 10.1056/NEJMra1808012
- Maurer M, Magerl M, Betschel S and others. ‘The international WAO/EAACI guideline for the management of hereditary angioedema—The 2021 revision and update‘. Allergy: European Journal of Allergy and Clinical Immunology 2022: volume 77, issue 7, pages 1,961–1,990. DOI: 10.1111/all.15214