Fibrodysplasia ossificans progressiva
Fibrodysplasia ossificans progressiva is an ultra-rare, progressive, life-limiting connective tissue disorder in which soft tissue and muscle turns to bone. This results in progressive immobility and difficulty with speaking, swallowing and breathing.
Overview
Fibrodysplasia ossificans progressiva (FOP) is characterised by congenital malformations of the great toes and progressive, episodic and abnormal development of bone in soft connective tissues (heterotopic ossification) in a characteristic pattern. Episodes can be triggered by minor trauma, including medical and dental procedures. FOP is caused by pathogenic variants in the ACVR1 gene, most often arising for the first time (de novo) in an affected individual.
Clinical features
Early diagnosis is important, to avoid unnecessary investigations and inappropriate treatments, which could exacerbate the condition. Early diagnostic signs, present at birth or in infancy, include:
- scalp nodules, which present in some affected individuals and typically regress spontaneously;
- neck stiffness due to abnormal cervical vertebrae; and/or
- congenital malformations of the great toes (hallux valgus); noting that
- at birth, all individuals with classic FOP have hallux valgus;
- the most common malformation is a shortened great toe with a malformed distal first metatarsal, and a missing or abnormal first phalanx (see figure 1); and
- congenital hallux valgus is also seen in individuals with BMPR1B-associated dysplasia.
Further clinical features include flare-ups, respiratory complications, skeletal associations and non-skeletal associations. These are described below.
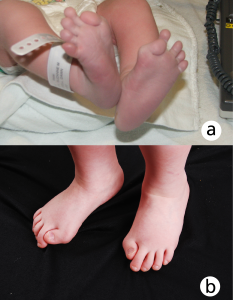
Figure 1: Congenital bilateral hallux valgus malformation in a) an infant and b) a child with FOP. Note the short, malformed and in-turned halluces. Photograph used with permission from the International Fibrodysplasia Ossificans Progressiva Association.
Flare-ups
Flare-ups are episodic painful soft-tissue swellings (sometimes called ‘flares’), often triggered by minor trauma. They typically begin by 10 years of age (see figure 2). Most flare-ups result in early-onset abnormal bone development in soft connective tissues, such as ligaments, tendons and skeletal muscle (heterotopic ossification); this may manifest as bony swellings. Flare-ups and heterotopic ossification may also occur spontaneously. Flare-ups affecting the hip may be confused with developmental dysplasia of the hip.
The diaphragm, tongue, extra-ocular muscles, cardiac muscle and smooth muscle are spared from flare-ups.
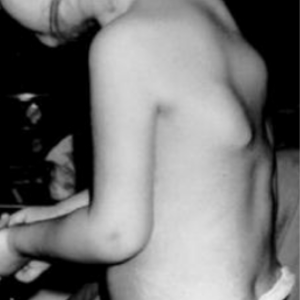
Figure 2: Characteristic swellings, on a child’s back, in a flare-up of FOP. Photograph used with permission of the International Fibrodysplasia Ossificans Progressia Association.
Respiratory complications
Respiratory complications associated with FOP include chest wall disease and kyphoscoliosis. These can result in thoracic insufficiency syndrome, which is the predominant cause of death in FOP patients. Restrictive lung disease is apparent in childhood and makes FOP life limiting, with the median age of death reported as 40 years.
Skeletal associations
In affected joints, clinical features include:
- progressive stiffness;
- degeneration; and
- ankylosis (fusion).
This usually progresses from the axial skeleton (cranium, thoracic cage and vertebral column) to the appendicular skeleton (shoulder girdle, pelvic girdle and upper and lower limbs) in the following order: neck, back, shoulders, elbows, hips, knees, wrists, ankles and then jaw.
Complications here include:
- progressive immobility, where most affected individuals depend on the use of a wheelchair by 30 years of age; and/or
- ankylosis of the jaw, which may compromise swallowing and speaking.
Other skeletal features include:
- short malformed thumbs;
- clinodactyly (curved finger);
- osteochondromas, most commonly in the proximal medial tibia;
- cervical spine fusions;
- scoliosis;
- temporomandibular joint malformation;
- short broad femoral neck;
- atypical distal limb reduction defects, affecting the fingers, in non-classical FOP – these may be misdiagnosed as an amniotic band defect or brachydactyly syndrome; and/or
- increased fracture risk, due to increased risk of falls and corticosteroid-related osteopenia.
Non-skeletal associations
These include:
- increased risk of renal stones;
- conductive hearing loss (common in FOP, so screening in childhood is recommended);
- gastrointestinal symptoms (can be associated with FOP); and
- lymphoedema.
Genomics
FOP is caused by heterozygous gain-of-function pathogenic variants in the ACVR1 gene. Only missense variants in specific domains have been recognised as causative and these lead to overactive bone morphogenic protein (BMP) and Activin A signalling, mediated by ACVR1.
Classic FOP, which affects most individuals with the condition, is caused by the substitution of the amino acid arginine for histidine at position 206 in the ACVR1 protein (p.Arg206His). Other variants in ACVR1 have been identified in a small proportion of individuals with FOP.
Some variants are associated with a milder disease course, while some are additionally associated with severe reduction deformities of the digits.
Diagnosis
Early recognition and clinical diagnosis of FOP allows the education of families to the risk of progressive heterotopic ossification. It also highlights to the patient and family the importance of avoiding exacerbating factors. This can include minor soft-tissue trauma such as intramuscular immunisations, soft tissue injuries, joint overuse, dental and surgical procedures, and viral illnesses.
A clinical diagnosis of FOP should be suggested in individuals with clinical features as described above. Though radiographic imaging isn’t required for diagnosis, recording specific changes in the halluces and extraosseous bone formation in affected areas would be supportive.
Genomic testing of FOP will look for a heterozygous pathogenic/likely pathogenic variant in the ACVR1 gene – ACVR1 testing is included in R104 Skeletal dysplasia – and the presence of this confirms a FOP diagnosis. (A saliva sample can be sent for genomic testing to avoid unnecessary blood sampling.) For more information about genomic testing, see Presentation: Child with suspected skeletal dysplasia.
Inheritance and genomic counselling
FOP is estimated to affect 1 in 1 million people worldwide and has been reported in diverse populations. Most cases of FOP arise sporadically because of a new (de novo) causative variant occurring in the egg or sperm. Rarely, an individual with FOP will have an affected parent.
FOP is inherited in an autosomal dominant pattern, with complete penetrance in classical FOP (meaning that all individuals with the causative variant in ACVR1 have signs and symptoms of the condition).
Individuals affected by an autosomal dominant condition have one working copy of the gene, and one with a pathogenic variant. The chance of a child inheriting the gene with the variant from an affected parent is 1 in 2 (50%). Pre-conception counselling should be offered. Prenatal testing and preimplantation genetic testing are available.
Pregnancy in affected females is rare and potentially life threatening to both mother and baby. Pregnancy should be managed in a high-risk centre following established guidelines, such as those by the International Clinical Council on Fibrodysplasia Ossificans Progressiva (see ‘Resources’, below).
Genomic counselling provided by a clinical genetics department is recommended for affected individuals. In de novo cases, clinical assessment/testing of siblings should still be considered due to the low risk of germline (gonadal) mosacism.
Management
There is currently no cure for FOP. Management of affected children and adults is complex and should be delivered via a multidisciplinary team, with referral to a specialist service with expertise in FOP. International FOP treatment guidelines are available, which also include emergency management guidelines.
The key aspects of management include:
- early diagnosis, with education about FOP for affected families and local healthcare providers;
- minimising the risk of trauma and joint overuse;
- avoiding iatrogenic harm, such as intramuscular injections and surgery;
- reducing risk of respiratory infections, for instance avoid the intranasal flu immunisation but give the intramuscular immunisation subcutaneously;
- maintaining good dental hygiene;
- the symptomatic management of flare-ups; and
- seeking occupational therapy input and providing mobility aids may be of help, but be careful to avoid joint overuse.
For information about clinical trials of potential new therapies for FOP, see the ClincalTrials.gov resource.
Resources
For clinicians
- GeneReviews: Fibrodysplasia Ossificans Progressiva
- Genomics England: NHS Genomic Medicine Service (GMS) Signed Off Panels Resource
- International Clinical Council on Fibrodysplasia Ossificans Progressiva: FOP Treatment Guidelines
- International FOP Association (IFOPA)
- IFOPA: FOP Registry
- National Organization for Rare Disorders: Fibrodysplasia ossificans progressiva
- NHS England: National Genomic Test Directory
- U.S. National Library of Medicine: ClinicalTrials.gov (database)
References:
- Valer JA, Sánchez-de-Diego C, Pimenta-Lopes C and others. ‘ACVR1 function in health and disease‘. Cells 2019: volume 8, issue 11, article number 1,366. DOI: 10.3390/cells8111366
For patients
- FOP Friends (UK charity providing patient information and support)
- International Fibrodysplasia Ossificans Progressiva Association (Charity providing patient information and support)