Epidermolysis bullosa
Epidermolysis bullosa is an umbrella term for a range of genetic conditions characterised by fragile skin that blisters easily following minor trauma. Severe subtypes can cause extensive blistering at birth, with affected neonates needing multidisciplinary care and having high morbidity.
Overview
Epidermolysis bullosa (EB) is a group of rare inherited genetic conditions that cause mechanical fragility of the skin and mucosal tissues. It can present at birth or in early childhood, with symptoms ranging from mild blistering to severe complications such as scarring, skin cancer and recurrent infections. EB is caused by pathogenic variants in genes responsible for skin barrier integrity, and can follow autosomal dominant or recessive inheritance patterns. Gene therapies for EB are in clinical use in some countries.
Clinical features
Clinical features of EB include:
- fragile skin prone to blistering and erosions;
- blisters and erosions on the hands, feet and other pressure-prone areas;
- scarring in some types of EB, leading to fusion of digits;
- blisters in mucosal tissues (such as the mouth and the oesophagus); and
- increased risk of squamous cell carcinoma in some types of EB.
Genomics
EB is caused by pathogenic variants in genes encoding structural proteins of the skin (see figure 1), such as COL7A1, KRT5 and KRT14. These pathogenic variants disrupt the skin’s ability to withstand mechanical stress, leading to blister formation.
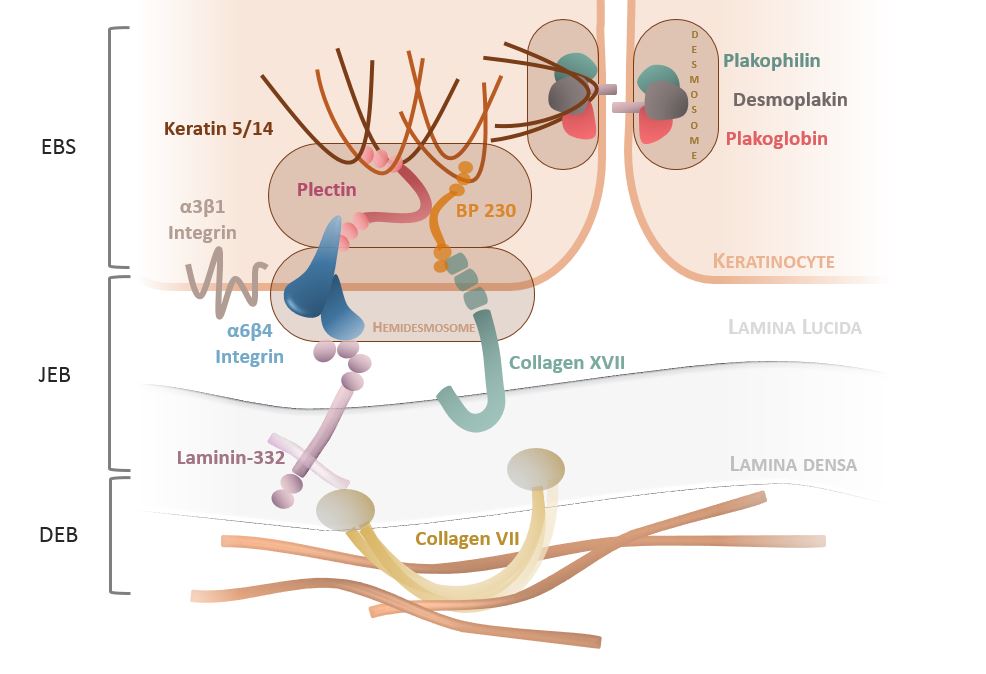
Figure 1: The dermo-epidermal junction in a case of epidermolysis bullosa
Image reproduced with kind permission from Dr Christine Prodinger.
Diagnosis
Diagnosis of EB involves:
- clinical evaluation of blistering patterns and severity;
- skin biopsy with immunofluorescence mapping; and/or
- genomic testing with a panel of relevant genes, which is used to identify causative pathogenic variants, confirm the diagnosis and guide family planning.
For information about genomic testing for suspected epidermolysis bullosa, see Neonate with skin blistering and fragility.
There are four designated EB centres of excellence (highly specialised services) around the UK and new patients and families should be referred to these centres to co-ordinate the care of suspected cases. They are:
- Great Ormond Street Children’s Hospital in London (children);
- Birmingham Women’s and Children’s Hospital (children);
- Guy’s and St Thomas’ Hospital in London (adults); and
- Solihull Hospital (adults).
Inheritance and genomic counselling
Severe forms of EB may affect about 1 in 50,000 live births globally. The condition can follow autosomal dominant or autosomal recessive inheritance patterns, depending on the subtype.
For autosomal dominant subtypes:
- individuals have one working copy of the gene, and one with a pathogenic variant;
- the chance of a child inheriting the gene with the variant from an affected parent is 1 in 2 (50%); and
- incomplete penetrance can occur, which means that not everyone who has the variant develops the disease.
For autosomal recessive subtypes:
- if both parents are carriers, with each pregnancy there is a:
- 1-in-4 (25%) chance of a child inheriting both gene copies with the pathogenic variant and therefore being affected;
- 1-in-2 (50%) chance of a child inheriting one copy of the gene with the pathogenic variant and one normal copy, and therefore being a healthy carrier; and
- 1-in-4 (25%) chance of a child inheriting both normal copies and being neither affected nor a carrier.
Genomic counselling is essential to discuss recurrence risks, carrier testing and implications for family members.
Management
Management focuses on wound care, infection prevention and pain management, and should be co-ordinated with an EB centre of excellence (listed above). Emerging therapies, such as gene therapy, are now in clinical use in some countries but are not yet widely available.
Referral of affected patients to clinical genetics should be considered to discuss onward management, family planning implications and cascade testing of relatives at risk.
Resources
For clinicians
- DEBRA: Healthcare professionals
- Genomics England: NHS Genomic Medicine Service (GMS) Signed Off Panels Resource
- Medizin Online: News about Epidermolysis Bullosa
- NHS England: National Genomic Test Directory
For patients
- British Association of Dermatologists: Epidermolysis bullosa simplex
- DEBRA