Agammaglobulinaemia types 1-10
Agammaglobulinaemia is a primary B-cell immunodeficiency characterised by susceptibility to infections, low or absent serum immunoglobulins and low or absent B cells.
Overview
Agammaglobulinaemia is a humoral immunodeficiency with reduced or absent antibody production. It is characterised by recurrent bacterial infections, with clinical features indistinguishable from X-linked agammaglobulinaemia. Presentation is typically in infancy, when the protective effect of maternal immunoglobulins wanes. Agammaglobulinaemia types 1–10 are genetically heterogenous conditions caused by autosomal dominant or recessive pathogenic variants in genes critical in early B-cell development.
Clinical features
Clinical features of agammaglobulinaemia include:
- increased susceptibility to extracellular bacterial infections, typically upper and lower respiratory tract infections;
- increased susceptibility to mycoplasma and giardia infections;
- severe or prolonged enteroviral infections;
- early onset neutropenia, which typically improves with time;
- bronchiectasis;
- chronic diarrhoea and colitis; and
- rarely, autoimmune manifestations such as arthritis and hypothyroidism.
Affected patients typically present in the first year of life, once maternal transplacental IgG have declined, usually six to eight weeks after birth. The first presentation may be a severe, life-threatening infection, such as pneumonia, empyema, meningitis, sepsis, cellulitis or septic arthritis.
Genomics
Agammaglobulinaemia types 1–10 are genetically heterogeneous conditions caused by pathogenic variants in the following genes (see table 1).
Table 1: The genes, proteins and modes of inheritance linked with agammaglobulinaemia types 1–10
| Gene | Protein | Inheritance | |
| Type 1 | IGHM | μ heavy chain | Autosomal recessive |
| Type 2 | IGLL1 | λ5 chain | Autosomal recessive |
| Type 3 | CD79A | Ig-α | Autosomal recessive |
| Type 4 | BLNK | B cell linker | Autosomal recessive |
| Type 5 | LRRC8 | volume regulated anion channel | Autosomal dominant |
| Type 6 | CD79B | immunoglobulin-β | Autosomal recessive |
| Type 7 | PIK3R1 | phosphatidylinositol 3-kinase (p110δ) | Autosomal dominant |
| Type 8 | TCF3 | E2A transcription factor | Autosomal recessive and dominant |
| Type 9 | SLC39A7 | zinc transporter ZIP7 | Autosomal recessive |
| Type 10 | SPI1 | PU.1 transcription factor | Autosomal dominant |
In boys, molecular investigation of suspected agammaglobulinaemia should start with genetic analysis of BTK, which accounts for 85% of cases (see Figure 1). In girls, and in boys in whom X-linked agammaglobulinaemia has been ruled out, recessive and dominant causes of agammaglobulinaemia should be considered.
Figure 1: Errors in B-cell development
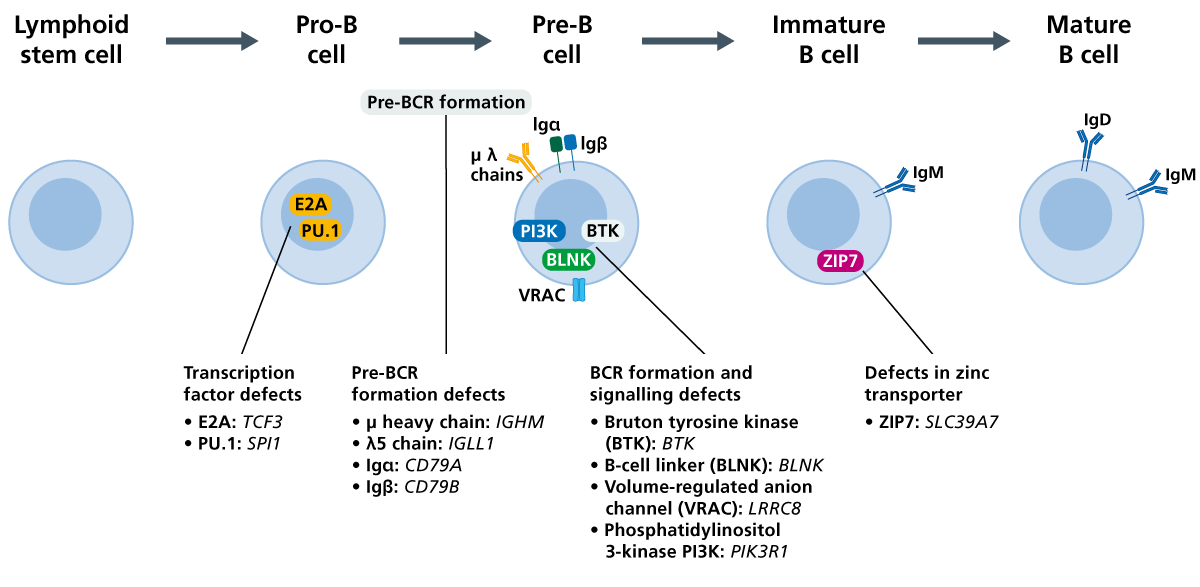
Pathogenic variants in genes essential for B-cell development are the cause of agammaglobulinaemia. (Click to enlarge in a new tab.)
Diagnosis
Agammaglobulinaemia can be diagnosed if:
- there is a history of recurrent infections before five years of age; and
- there are fewer than 2% circulating B cells (CD19 and CD20), preferably in two separate determinations, and a normal number of T cells (CD3, CD4, and CD8); and
- serum IgG levels are below:
- 200mg/dl in infants aged <12 months;
- 500mg/dl in children aged >12 months; or
- there are normal IgG levels, with IgA and IgM two standard deviations (2SD) below the mean.
For more information, see the European Society for Immunodeficiencies’ diagnostic criteria.
Molecular confirmation of agammaglobulinaemia types 1–10 is made through genetic analysis, with the finding of pathogenic or likely pathogenic variants in the above genes.
Agammaglobulinaemia may be identified before any symptoms appear, for example through the Generation Study. Confirmation of the diagnosis will require referral to clinical immunology services. Please refer to the local pathway for your region for this condition.
Inheritance and genetic counselling
Agammaglobulinaemia can be autosomal dominant, autosomal recessive or X-linked. This article focuses on the autosomal dominant and recessive forms.
- If both parents are carriers of an autosomal recessive condition, with each pregnancy there is a:
- 1-in-4 (25%) chance of a child inheriting both gene copies with the pathogenic variant and therefore being affected;
- 1-in-2 (50%) chance of a child inheriting one copy of the gene with the pathogenic variant and one normal copy, and therefore being a healthy carrier themselves; and
- 1-in-4 (25%) chance of a child inheriting both normal copies and being neither affected nor a carrier.
- Individuals affected by an autosomal dominant condition have one working copy of the gene, and one with a pathogenic variant.
- The chance of a child inheriting the gene with the variant from an affected parent is 1 in 2 (50%).
- Incomplete penetrance can occur (that is, not everyone who has the variant develops the disease).
Note that de novo variants can also occur, and that reproductive options are available for couples at risk of having an affected child. Genetic counselling is recommended.
When a patient receives a diagnosis, careful consideration should be given to other family members who may also be affected. A family history should be taken and, where appropriate, genomic testing for the familial variant/s should be offered.
If you are discussing genomics concepts with your patients, you may find it helpful to use the visual communication aids for genomics conversations.
Management
The mainstay of treatment is immunoglobulin replacement therapy. This is available in many different preparations, and can be administered intravenously or subcutaneously. Prophylactic antibiotics may also be used to prevent infections, and prompt treatment with antibiotics for a longer course is required when the patient develops an infection.
Agammaglobulinaemia may be identified before any symptoms appear, for example through the Generation Study. Management of these individuals may differ from those presenting symptomatically.
More information about management can be found in the resources section below.
Resources
For clinicians
- European Society for Immunodeficiencies: Diagnostic criteria
- NHS England: Clinical commissioning criteria policy for use of therapeutic immunoglobulin (Ig) England 2025 (PDF, 37 pages)
References:
- Abolhassani H, Parvaneh N, Rezaei N and others. ‘Genetic defects in B-cell development and their clinical consequences’. Journal of Investigational Allergology & Clinical Immunology 2014: volume 24, issue 1, pages 6–22 (PDF, 17 pages)
- Conley ME, Broides A, Hernandez-Trujillo V and others. ‘Genetic analysis of patients with defects in early B-cell development‘. Immunological Reviews 2005: volume 203, issue 1, pages 216–234. DOI: 10.1111/j.0105-2896.2005.00233.x