Acrodermatitis enteropathica
Acrodermatitis enteropathica is a treatable genetic condition causing intermittent skin and bowel symptoms.
Overview
Acrodermatitis enteropathica (AE) arises due to an inherited zinc transporter defect, resulting in reduced intestinal absorption of zinc. Zinc deficiency results in alopecia, diarrhoea, bullous skin lesions and failure to thrive. It is caused by autosomal recessive variants in the SLC39A4 gene. These variants cause disturbances in the uptake, transport and overall homeostasis of zinc, with disturbed function of enzymes requiring zinc as a co-factor leading to clinical symptoms.
Clinical features
The initial presentation of AE is commonly a characteristic dermatitis, classically manifesting with acral and perioral distribution (figure 1).
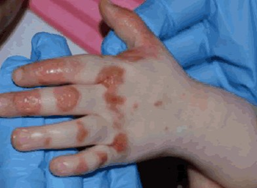
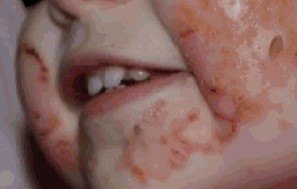
Figure 1: Acral and perioral acrodermatitis enteropathica lesions
Images reproduced from Sutton J and Newin T, 2016 (see ‘Resources’ section for full reference) under Creative Commons licensing.
Other features include:
- sharp demarcation of dermatitis relative to normal skin;
- horseshoe or U-shaped peri-orofacial distribution, with lip sparing;
- red and scaly plaques, which can evolve to become vesicles, bullae, pustules or blisters;
- glossitis, mouth ulcers and angular cheilitis;
- perianal excoriations and rash on the buttocks; and
- involvement of extensor surfaces of the knees or elbows.
Skin lesions are prone to secondary infection. Nails can be soft and have a ridged texture. Alopecia affects the scalp, eyebrows and eyelashes.
Non-dermatological symptoms include:
- photophobia and blepharoconjunctivitis;
- diarrhoea, loss of appetite and weight loss;
- depression, low mood and emotional dysregulation in older children and adults, or irritability in infants;
- failure to thrive;
- hearing loss;
- amenorrhea in females; and
- hypogonadism in males.
The clinical presentation of AE can be heterogeneous, occurring early in infancy in formula-fed babies and weaning breast-fed babies due to higher bioavailability of zinc in breast milk. AE can also present later in childhood, or as late as adulthood in some patients.
Genomics
AE is an autosomal recessive condition caused by homozygous or compound heterozygous variants in the SLC39A4 gene on chromosome 8q24.
Diagnosis
Diagnosis of AE can be made in different ways. Some of the presenting symptoms are non-specific, and as this is a rare condition many clinicians will not be aware of it. In the UK, the National Genomic Test Directory has several clinical indications that include the SLC39A4 gene:
- R164 Epidermolysis bullosa and congenital skin fragility;
- R331 Intestinal failure or congenital diarrhoea;
- R332 Rare genetic inflammatory skin disorders;
- R98 Likely inborn error of metabolism; and
- R27 Severe paediatric disorders.
When there is a clinical suspicion of AE, the following biochemical features support the diagnosis:
- low zinc levels in serum, urine and/or hair;
- low serum alkaline phosphatase; and
- low haemoglobin and leucocyte levels on full blood count.
However, results are not always reliable.
There is a zinc absorption test available, but it is cumbersome to use. Some clinicians will instead trial a zinc supplementation regime, which results in a rapid and dramatic clinical improvement in affected patients.
If there is a clinical or histological suspicion of the condition, genomic testing can confirm the diagnosis.
AE may be identified before any symptoms appear if the patient is a participant in the Generation Study. Confirmation of the diagnosis will require referral to local clinical genetic and/or metabolic services. Please refer to your local pathway.
Inheritance and genomic counselling
AE is an autosomal recessive condition, with a prevalence of 1 to 9:1,000,000, independent of ethnicity or sex.
If both parents are carriers of an autosomal recessive condition, with each pregnancy there is a:
- 1-in-4 (25%) chance of a child inheriting both gene copies with the pathogenic variant and therefore being affected;
- 1-in-2 (50%) chance of a child inheriting one copy of the gene with the pathogenic variant and one normal copy, and therefore being a healthy carrier of the condition; and
- 1-in-4 (25%) chance of a child inheriting both normal copies and being neither affected nor a carrier.
If parents are concerned about a recurrence risk, they can be referred for genetic counselling. Reproductive options may be available, including testing in early pregnancy or preimplantation genomic testing.
Consideration should be given to the siblings of patients with AE, as they are at a higher theoretical risk. If a genetic diagnosis has been made, testing for the familial variant is available.
Management
The prognosis of AE is generally good with timely diagnosis and zinc replacement. The condition can be potentially fatal without appropriate treatment.
- Initial treatment of AE is with high-dose oral zinc supplementation, but there is potential for toxicity with long-term treatment.
- Dietary advice is essential to correct and prevent future potential inadequate intake.
- Serum zinc levels should be taken for adjustment of oral supplements to the lowest effective dose.
Resources
For clinicians
- DermNet: Acrodermatitis enteropathica
- OMIM: #201100 Acrodermatitis enteropathica
- StatPearls: Acrodermatitis enteropathica
References:
- Abu-Duhier F, Pooranachandran V, McDonagh AJG and others. ‘Whole genome sequencing in an acrodermatitis enteropathica family from the Middle East‘. Dermatology Research and Practice 2018. DOI: 10.1155/2018/1284568
- Corbo MD and Lam J. ‘Zinc deficiency and its management in the pediatric population: A literature review and proposed etiologic classification‘. Journal of the American Academy of Dermatology 2013: volume 69, issue 4, pages 616–624. DOI: 10.1016/j.jaad.2013.04.028
- Horcadaja-Reales C, Barchino-Ortiz L, Conde-Montero E and others. ‘Acrodermatitis enteropathica in an adult‘. Australian Family Physician 2015: volume 44, issue 5, pages 299–300. PMID: 26244999
- Küry S, Dréno B, Bézieau S and others. ‘Identification of SLC39A4, a gene involved in acrodermatitis enteropathica‘. Nature Genetics 2002: volume 31, issue 3, pages 239–40. DOI: 10.1038/ng913
- Sutton J and Newin T. ‘A case of acrodermatitis enteropathica‘. Journal of Clinical & Experimental Dermatology Research 2016: volume, issue 2, page 329. DOI:10.4172/2155-9554.1000329 (PDF, five pages)
For patients
- Genetic and Rare Diseases Information Center: Acrodermatitis enteropathica
- Metabolic Support UK: Acrodermatitis enteropathica
- National Organization for Rare Disease: Acrodermatitis enteropathica
- The Autoimmune Association